阿司匹林双嘧达莫片的含量测定及含量均匀度研究
2019-09-16田海燕崔晓雨刘洪海
李 宇,田海燕,崔晓雨,刘洪海△
(1.德州学院职业教育学院,山东 德州 253023; 2.山东省德州市食品药品检验检测中心,山东 德州 253015)
阿司匹林双嘧达莫片为肠溶衣与薄膜衣的双层包衣片,内层含阿司匹林,外层含双嘧达莫。原标准[1]采用滴定分析法测定阿司匹林的含量,分光光度法测定双嘧达莫的含量。实际工作中,剥离外层(双嘧达莫片)时存在迸溅损失,以及迸溅时不确定损失的是主药成分还是包衣材料,且试验时间太长,吸湿及刮片也影响内外层的含量测定结果。新标准[2]采用高效液相色谱法测定其含量及含量均匀度,但色谱分析耗时较长。本研究中参考有关标准及文献[2-4],建立了紫外分光光度法测定阿司匹林双嘧达莫片的含量及含量均匀度,现报道如下。
1 仪器与试药
Tu-1901型双光束紫外可见分光光度计(北京通用仪器有限责任公司);XS105 DualRange型电子分析天平(瑞士Mettler Toledo公司);Agilent 1260型高效液相色谱仪,含G7114A型紫外检测器(美国安捷伦科技有限公司)。
阿司匹林对照品(含量99.8%,批号为100113-201405),双嘧达莫对照品(含量 99.8%,批号为100244-201003),水杨酸对照品(含量99.3%,批号为100106-201605),均购自中国食品药品检定研究院。乙腈为色谱纯,冰乙酸和四氢呋喃为优级纯,其余均为分析纯。
阿司匹林双嘧达莫片,共3批,规格均为每片含阿司匹林75 mg、双嘧达莫25 mg,分别购自德州博诚制药有限公司(样品1,批号为A20171202)、湖北百科亨迪药业有限公司(样品2,批号为171002)、湖北百科亨迪药业有限公司(样品3,批号为171003)。
2 方法与结果
2.1 测定波长选择
取双嘧达莫对照品适量,用0.1 mol/L盐酸溶液溶解,并稀释成质量浓度为10 μg/mL的溶液,在200~300 nm波长范围内扫描,结果在233 nm和284 nm波长处有最大吸收,故以284 nm为双嘧达莫的测定波长。
取阿司匹林对照品适量,加少量乙醇溶解,用pH 6.8磷酸盐缓冲液稀释成质量浓度为150 μg/mL的溶液,在250~300nm波长范围内扫描,结果在266nm波长处有最大吸收,故以266 nm为阿司匹林的测定波长。
参照双嘧达莫层和阿司匹林层,分别将相应辅料用0.1 mol/L盐酸溶液和pH 6.8磷酸盐缓冲液溶解,并稀释成相应浓度,滤过,续滤液分别在200~300 nm和250~300 nm波长处扫描,在相应的吸收波长284 nm和266 nm处无吸收,不干扰主成分的含量测定。
2.2 线性关系考察
双嘧达莫:取双嘧达莫对照品10 mg,精密称定,置50 mL容量瓶中,用0.1 mol/L盐酸溶液溶解并定容。分别吸取 1.0,2.0,3.0,5.0,6.0,7.0,8.0 mL,置 50 mL容量瓶中,用0.1 mol/L盐酸溶液定容。在284 nm波长处测定吸光度(A),以质量浓度(C,μg/mL)对A进行线性回归,得回归方程C=14.884A+0.278 6,r=0.999 7(n=7)。结果表明,双嘧达莫质量浓度在4~32 μg/mL范围内与吸光度线性关系良好。
阿司匹林:取阿司匹林对照品30 mg,精密称定,置50 mL容量瓶中,用少量乙醇溶解,加pH 6.8的磷酸盐缓冲液定容。分别吸取 2.0,3.0,4.0,5.0,6.0,7.0,8.0 mL,置20 mL容量瓶中,加pH 6.8的磷酸盐缓冲液定容,在266 nm波长处测定吸光度(A),以质量浓度(C,μg/mL)对A进行线性回归,得回归方程C=327.79A-4.081,r=0.999 8(n=7)。结果表明,阿司匹林质量浓度在60~240 μg/mL范围内与吸光度线性关系良好。
2.3 含量测定
2.3.1 紫外分光光度法
取样品10片,置小烧杯中,加0.1 mol/L盐酸溶液适量,超声溶解双嘧达莫层,并转移至1 000 mL容量瓶中,用少量蒸馏水洗涤肠溶衣表面,洗液并入1 000 mL容量瓶中,用0.1 mol/L盐酸溶液定容,摇匀,滤过,取续滤液2.0 mL,置50 mL容量瓶中,加0.1 mol/L盐酸溶液定容,摇匀,照分光光度法,在284 nm波长处测定吸光度,按标准曲线法计算双嘧达莫标示量的百分含量。详见表1。
取上述测定双嘧达莫后所剩的具有完整肠溶衣的内层10片,置研钵中,研细,加少量乙醇溶解,用pH 6.8磷酸盐缓冲液转移至500 mL容量瓶中并定容,摇匀,滤过,取续滤液5.0mL,置50mL容量瓶中,用pH 6.8磷酸盐缓冲液定容,摇匀,照分光光度法在266 nm波长处测定吸光度,按标准曲线法计算占阿司匹林标示量的百分含量。详见表1。
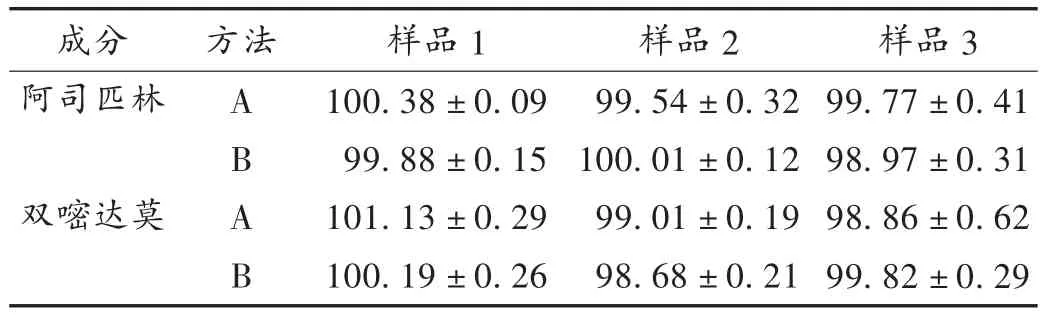
表1 样品标示量百分含量测定(%)
2.3.2 高效液相色谱法
取样品,采用 Agilent Zorbax SB C18柱(250 mm×0.6 mm,5 μm),以乙腈 -四氢呋喃 -冰乙酸 -水(20∶5 ∶5∶70,V/V/V/V)为流动相,柱温 30℃,流速为1.0 mL/min,检测波长 276 nm(阿司匹林)、284 nm(双嘧达莫)[2]。
阿司匹林:理论板数按阿司匹林峰计为14 514,阿司匹林峰保留时间为6.6 min,阿司匹林峰与水杨酸峰及双嘧达莫峰的分离度分别为5.0和19.6。含量测定结果见表1。
双嘧达莫:理论板数按双嘧达莫峰计为5 401,双嘧达莫峰保留时间为5.3 min,双嘧达莫峰与阿司匹林峰的分离度为4.9。含量测定结果见表1。
2.4 稳定性试验
取质量浓度约为10 μg/mL的双嘧达莫样品溶液,室温下放置 0,1.0,2.0,3.0,4.0 h,测定吸光度,结果的RSD为0.11%(n=5);取质量浓度约为150 μg/mL的阿司匹林样品溶液,室温下放置 0,0.5,1.0,1.5,2.0 h,测定吸光度,结果的RSD为0.39%(n=5)。结果表明,2种样品溶液室温下在测试时间内稳定。
2.5 回收率试验
取阿司匹林和双嘧达莫对照品,分别按标示量的80%,100%,120%配制内层和外层辅料的混合物各3份,用含量测定方法的溶剂分别配制成相应质量浓度的供试液,依法测定,结果见表2。
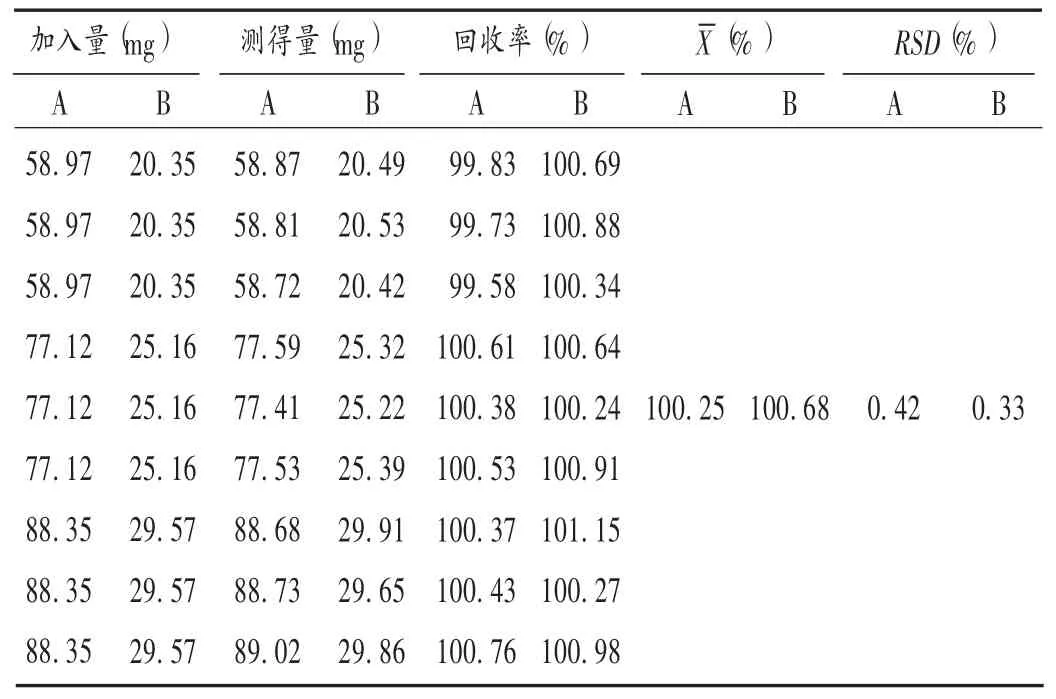
表2 加样回收试验结果(n=9)
2.6 双嘧达莫含量均匀度测定
3批样品每批均取10片,分别置10个小烧杯中,加0.1 mol/L盐酸溶液适量,分次超声溶解外层(双嘧达莫层),并转移至100 mL容量瓶中,依法测定双嘧达莫含量,得各片含量并计算3批样品的A+2.2S分别为6.1,7.2,6.7,符合规定,表明含量均匀度较好。
3 讨论
参照有关标准[2,5],建议用2.6项下10份样品平均标示量的百分含量作为含量的测定值,更易观察每片双嘧达莫层的溶解情况。新标准[2]虽然解决了外层双嘧达莫测定时刮片引起的误差,但依然存在测定阿司匹林时刮片的费时费力问题。测阿司匹林含量时所用肠溶衣内层应完好无损,无裂缝、崩解或软化现象。
本研究中还考察了高效液相色谱法,测定结果与紫外分光光度法基本一致。也考察了标准所允许游离水杨酸的最大限量,对结果无影响。紫外分光光度法未采用对人体有害的有机溶剂乙腈、四氢呋喃等,健康环保,且快速准确,可作为测定阿司匹林双嘧达莫片含量及含量均匀度的方法。
