HPLC法测定青蒿素哌喹片中青蒿素有关物质
2019-09-12张志坚杨兆丽谢丹娥刘澎张加颂宋健平
张志坚, 杨兆丽, 谢丹娥, 刘澎, 张加颂, 宋健平
(1.广东新南方青蒿药业股份有限公司,广东梅州 514300;2.广东新南方青蒿科技有限公司,广东梅州 514300;3.广州中医药大学,广东广州 510405)
青蒿素哌喹片为青蒿素和哌喹组成的复方制剂,用于治疗恶性疟、间日疟和三日疟。该药问世于2006年,属于国家一类新药,具有自主知识产权,是广东新南方青蒿药业股份有限公司独家生产品种。青蒿素哌喹片原标准为国家食品药品监督管理局标准YBH08022006,现行标准为《中国药典》(二部)“青蒿素哌喹片”项下标准[1],采用高效液相色谱(HPLC)法检查有关物质,但该法未能将哌喹与被检成分完全分离,灵敏度和专属性较差。因此,为严格控制药品质量,本单位决定优化质量标准,提高青蒿素有关物质的灵敏度、专属性和可靠性,本课题组参考2015年版《中国药典》、《国际药典》第3版[2]中“青蒿素”及其他文献[3-6]中有关青蒿类有关物质的测定方法,研究HPLC法测定青蒿素哌喹片中青蒿素有关物质的方法,并建立青蒿素有关物质限度。现将研究结果报道如下。
1 仪器与试药
1.1仪器岛津LC-20A高效液相色谱仪。赛默飞Ultimate 3000高效液相色谱仪。色谱柱:YMCTriart C18柱(4.6 mm × 250 mm,5 μm);THERMOHypersil GOLD 柱(4.6 mm ×250 mm, 5 μm);Welch-Xtimate柱(4.6 mm×250 mm,5 μm)。XP204S型万分之一分析天平、XS205DV型十万分之一分析天平(美国梅特勒——托利多有限公司)。
1.2试药青蒿素哌喹片,广东新南方青蒿药业有限公司,批号:20141001、20141002、20141003;青蒿素,广东新南方青蒿药业有限公司,批号:Y13120001;哌喹,广东新南方青蒿药业股份有限公司,批号:20161030;杂质A(青蒿烯),Toronto Research Chemicals,批号:9-MMH-173-1;杂 质 B(9-epi-Artemisinin), Toronto Research Chemicals,批号:2-NKM-68-8;杂质I[7-氯-4-(1-哌嗪基)喹啉],中国药品检定研究院,批号:101339-201501;杂质II(7-氯-4-羟基喹啉),中国药品检定研究院,批号:101340-201501;杂质Ⅲ[1,4-二(7-氯喹啉-4-基)哌嗪],中国药品检定研究院,批号:101341-201501;甲醇、乙腈均为色谱级(上海安谱公司);水为超纯水;三氯乙酸(分析纯,国药集团);其余试剂均为分析纯。
2 方法与结果
2.1色谱条件及系统适用性试验色谱条件:色谱柱为YMC-Triart C18柱(250 mm× 4.6 mm,5 μm);流动相为乙腈-0.01%三氯乙酸(50∶50);流速为1.0 mL/min;检测波长为210 nm;柱温为35℃;进样量为20 μL;青蒿素峰与杂质A峰的分离度应大于4.0,相邻色谱峰间分离度应大于1.5。系统适用性溶液配制:分别精密称取青蒿素原料及其杂质A对照品、杂质B对照品适量,用乙腈溶解稀释,制成含青蒿素5 mg/mL、杂质A 7.5 μg/mL、杂质B 15 μg/mL的系统适用性溶液。
2.2测定方法取青蒿素哌喹片细粉适量(约相当于青蒿素50 mg),加乙腈溶解并稀释制成每1 mL中约含青蒿素5 mg的溶液,滤过,取续滤液作为供试品溶液。精密量取1 mL,置100 mL量瓶中,用乙腈稀释至刻度,摇匀,作为对照溶液。精密量取上述溶液各20 μL,分别注入液相色谱仪,按上述色谱条件测定,记录色谱图至主成分峰保留时间的1.5倍。
2.3专属性试验
2.3.1 杂质定位与分离试验 按处方比例配制供试品溶液,青蒿素及其杂质A、B对照品溶液,哌喹及其杂质Ⅰ、Ⅱ、Ⅲ对照品溶液,不含青蒿素的阴性对照溶液,按“2.1”项下色谱条件测定,各杂质与主峰、杂质与杂质间分离度均达到要求,阴性不干扰测定。其中5.7 min处色谱峰为未知杂质峰1,因其含量小于0.1%,故不需要对其进行鉴定研究。6.7 min处色谱峰为溶剂杂质峰,在空白溶剂试验中已经证实过。杂质A峰、杂质B峰的分离度为2.43,杂质B峰与青蒿素峰的分离度为3.56,均达到完全分离。色谱图见图1~3。
2.3.2 强制降解试验 取阴性(空白辅料+哌喹)、哌喹原料、青蒿素原料、按处方比例配制的供试品适量(约相当于青蒿素50 mg)置10 mL量瓶中,加入0.5 mol·L-1HCL溶液1 mL,室温放置5 min后,加入0.5 mol·L-1NaOH溶液1 mL,用乙腈稀释至刻度,摇匀,滤过,作为酸破坏样品。取阴性(空白辅料+哌喹)、哌喹原料、青蒿素原料、按处方比例配制的供试品适量(约相当于青蒿素50 mg)置10 mL量瓶中,加入0.5 mol·L-1NaOH溶液1 mL,室温放置5 min后,加入0.5 mol·L-1HCL溶液1 mL,用乙腈稀释至刻度,摇匀,滤过,作为碱破坏样品。取阴性(空白辅料+哌喹)、哌喹原料、青蒿素原料、按处方比例配制的供试品适量(约相当于青蒿素50 mg)置10 mL量瓶中,加体积分数30%过氧化氢2 mL,室温放置3 h后,用乙腈稀释至刻度,摇匀,滤过,作为氧化破坏样品。取阴性(空白辅料+哌喹)、哌喹原料、青蒿素原料、按处方比例配制的供试品适量(约相当于青蒿素50 mg)置10 mL量瓶中,加水2 mL后,90℃水浴2 h,放冷至室温,用乙腈稀释至刻度,摇匀,滤过,作为水溶液高温破坏样品。样品置105℃干燥3 h后,取阴性(空白辅料+哌喹)、哌喹原料、青蒿素原料、按处方比例配制的供试品适量(约相当于青蒿素50 mg)置10 mL量瓶中,加乙腈适量超声溶解后,用乙腈稀释至刻度,摇匀,滤过,作为高温破坏样品。样品在强光(照度为4 500±500 lx)下照射11 d后,取阴性(空白辅料+哌喹)、哌喹原料、青蒿素原料、按处方比例配制的供试品适量(约相当于青蒿素50 mg)置10 mL量瓶中,加乙腈适量超声溶解后,用乙腈稀释至刻度,摇匀,滤过,作为光照破坏样品。按“2.1”项下色谱条件测定,结果显示:各强制降解条件下空白溶液和空白辅料对本品制剂和原料药有关物质的检测均无干扰;各强制降解条件下原料药供试品溶液中主峰与各杂质峰、各主要杂质峰之间均能有效分离;各强制降解条件下制剂供试品溶液中主峰与各杂质峰、各主要杂质峰之间均能有效分离。酸、碱、氧化、水溶液高温、高温及强光降解条件下,青蒿素原料供试品溶液的物料平衡指数分别为100.4%、100.8%、100.1%、100.0%、101.9%、100.1%,制剂供试品溶液的物料平衡指数分别为97.8%、90.2%、99.0%、97.7%、99.4%、97.4%,青蒿素+阴性供试品溶液的物料平衡指数分别为100.6%、90.8%、98.8%、95.0%、103.7%、102.1%,均在90.0%~110.0%之间。表明该方法对各主要降解杂质均能较好检出,该法能有效地检出各破坏试验产生的杂质,且各杂质峰均能与主峰达到基线分离,专属性较好。
2.3.3 色谱峰归属 方法同“2.3.2”项下强制降解试验。根据降解试验结果,酸、碱、氧化、水溶液高温、高温及强光降解条件下青蒿素分别在5.497、5.510、5.043、5.500、16.410、5.703 min保留时间之后产生降解产物,说明青蒿素有关物质均在5 min之后产生。而哌喹在上述降解条件下在3~5 min之间均有降解产物产生。因空白辅料中哌喹为近期生产批次,不能完全模拟当时生产所用的空白辅料,在4 min左右的色谱峰无法解释,但从降解试验光谱图分析其化学结构,判断是哌喹的降解产物,而哌喹有关物质的研究在哌喹色谱条件下分析,在此条件下不再研究。为保证青蒿素有关物质检测的准确性,减少哌喹及其辅料的干扰,我们确定青蒿素有关物质的积分起始时间为4.5 min。
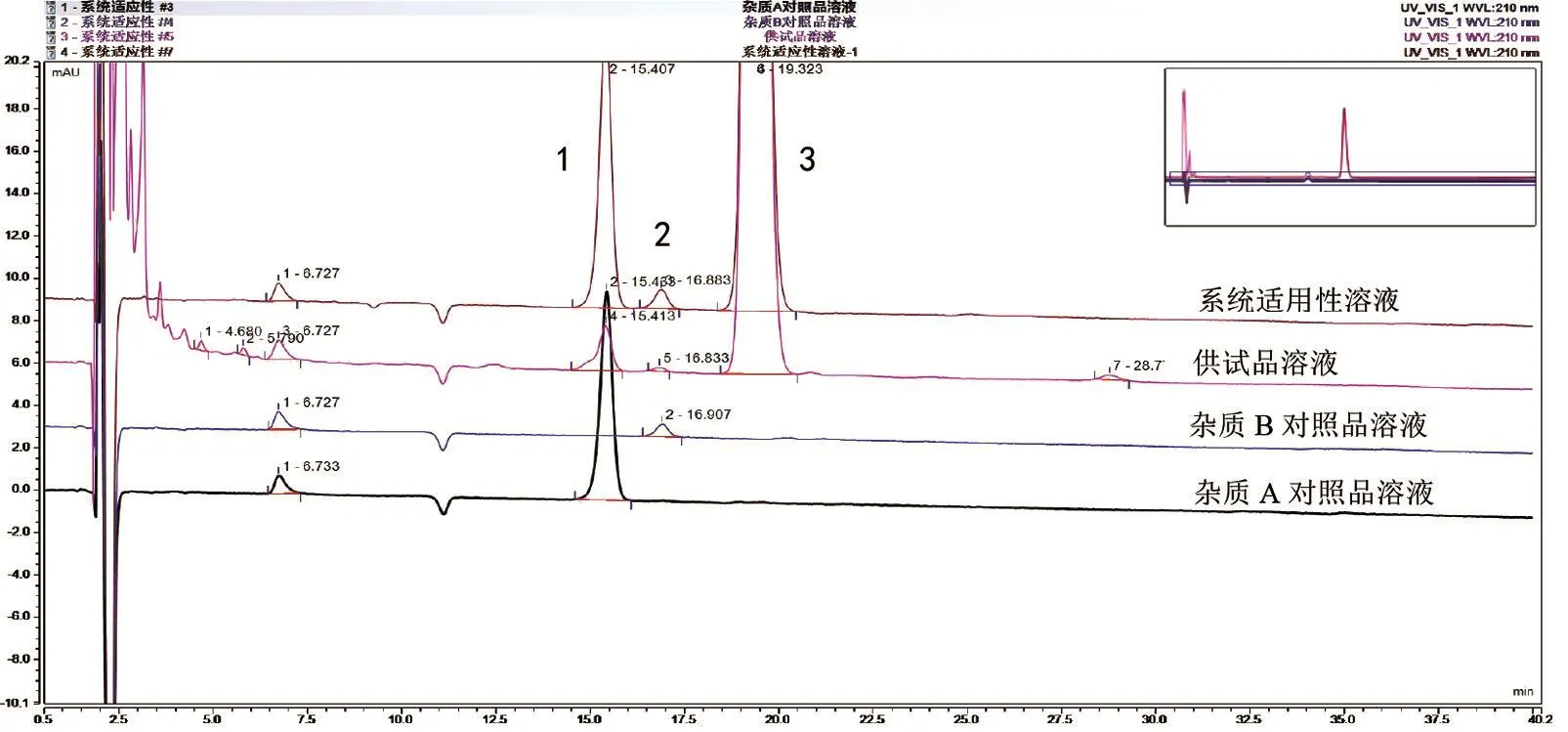
图1 系统适用性色谱图Figure 1 HPLC chromatogram of system suitability
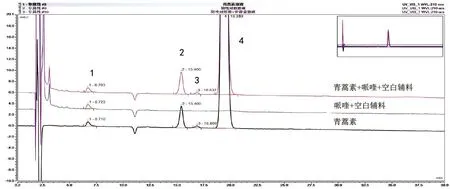
1:溶剂杂质;2:杂质A;3:杂质B;4:青蒿素
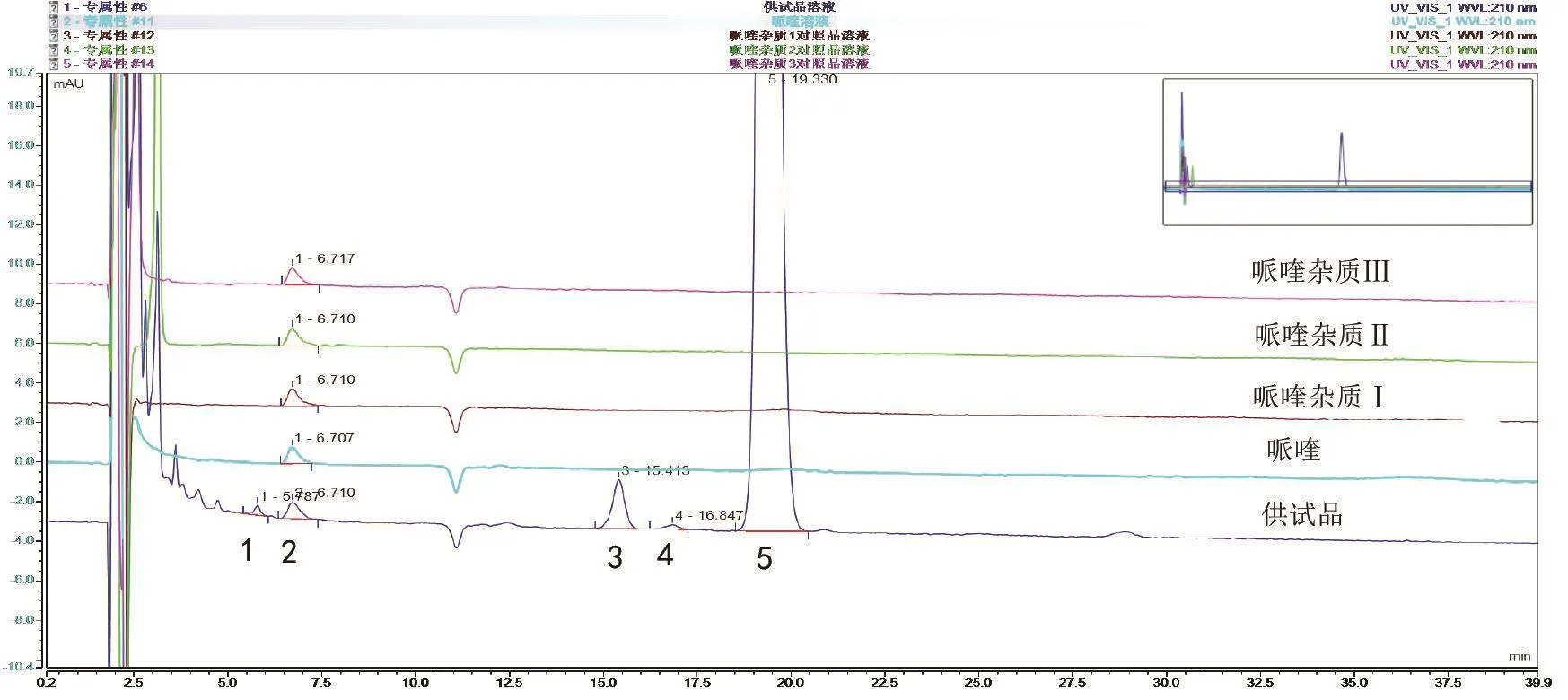
图3 杂质定位与分离试验色谱图(2)Figure 3 HPLC chromatogram of impurity location and separation test(2)
2.4检测限与定量限分别精密量取“2.3.1”项下青蒿素及其杂质A、B对照品溶液适量,用乙腈逐步稀释至各样品主峰信噪比为3∶1和10∶1,按“2.1”项下色谱条件测定。青蒿素的定量限和检出限分别为15、5 μg/mL,杂质A的定量限和检出限分别为0.25、0.075 μg/mL,杂质B的定量限和检出限分别为10、3 μg/mL。杂质A、B的最低检测限浓度均低于该杂质的报告限度浓度,反映该分析方法的灵敏度符合要求。
2.5线性关系及范围以杂质限度为100%,从定量限开始至杂质限度的200%~300%,分别取8个点,以浓度横坐标(X),峰面积为纵坐标(Y),进行线性回归分析。分别精密称量青蒿素及其杂质A、B对照品适量,用乙腈溶解并逐步稀释至青蒿素浓度依次为 15、20、25、30、35、40、45、50 μg/mL,杂质 A 浓度依次为 0.25、1.0、5.0、10.0、15、20、25、30 μg/mL,杂质B浓度依次为10、 15、 20、 25、 30、 35、 40、 45 μg/mL。 按“2.1”项下色谱条件测定,得到回归方程。青蒿素:Y=0.014 1 X+0.007 4,r=0.999 4;杂质A:Y=0.5164 X+0.045,r=0.999 9;杂质 B:Y=0.015 7 X-0.014 3,r=0.998 1。各线性方程的相关系数(r)均大于0.990,Y轴截距均在100%响应值的25%以内,响应因子的相对标准差均小于10%。结果表明青蒿素浓度在15~51 μg/mL范围内线性关系良好,杂质A浓度在0.3~30 μg/mL范围内线性关系良好,杂质B浓度在10~46 μg/mL范围内线性关系良好。
通过线性斜率比值计算得,杂质A校正因子为0.027,杂质B校正因子为0.90。参照青蒿素原料质量标准(国际药典和中国药典),确定杂质A校正因子为0.027,又因杂质B校正因子在0.9~1.1范围内,确定杂质B校正因子为1.00。
2.6加样回收率试验分别精密称取青蒿素哌喹片细粉适量加乙腈适量制成约相当于含青蒿素20 mg/mL的溶液,过滤后,精密量取2.5 mL,12份,分别置10 mL量瓶中。再分别精密量取对照品贮备液(含杂质A 15 μg/mL,杂质B 30 μg/mL)2.5、3.3、5、7.5 mL,用乙腈稀释至刻度,即得各浓度供试品溶液。浓度分别为供试品总量限度的50%、66%、100%、150%,即杂质A约相当于主成分的0.075%、0.100%、0.150%、0.225%,杂质B约相当于主成分的0.15%、0.20%(为杂质B定量限)、0.30%、0.45%,每个浓度平行制备3份平行样。按“2.1”项下色谱条件测定,计算加样回收率。杂质A的加样回收率(n=12)为96.6%,相对标准偏差(RSD,sR)为1.04%;杂质B的加样回收率(n=9)为85.9%,sR为3.21%。各浓度平均回收率均在80%~120%间,相对标准偏差在10%内。
2.7精密度试验按“2.1”项下系统适用性溶液和色谱条件,重复进样6次,计算杂质A、杂质B和青蒿素峰面积的sR为0.14%、3.39%、0.02%,符合精密度实验要求。
2.8重复性试验分别精密称取青蒿素哌喹片细粉适量(约相当于青蒿素50 mg),置10 mL量瓶中,精密移入0.1 mg/mL的杂质B对照品贮备液1.5 mL,加乙腈适量溶解,定容摇匀滤过,取续滤液作为供试品溶液。平行制备6份。按“2.1”项下色谱条件测定,计算杂质A、杂质B含量的sR为3.23%、4.18%,符合重复性实验要求。
2.9溶液稳定性试验按“2.1”项下系统适用性溶液和色谱条件,分别于0、4、8、12、24、48 h测定,记录色谱图。48 h内样品溶液中杂质A、杂质B和青蒿素峰面积的sR为0.41%(不超过10%)、3.68%(不超过10%)、0.64%(不超过2%),各峰面积均未出现显著性变化。结果符合验证要求,表明溶液在48 h内测定结果稳定。
2.10耐用性试验分别考察了不同色谱柱[YMCTriart C18(2)柱 、 THERMO-Hypersil GOLD 柱 ,Welch-Xtimate柱],乙腈-0.01%三氯乙酸流动相不同体积比(45∶55、50∶50、55∶45),不同流速(0.9、1.0、1.1 mL/min),不同检测波长(208、210、212 nm),不同三氯乙酸浓度(0.01%、0.05%、0.1%),不同柱温(33、35、37℃),不同品牌滤膜(津腾、安谱、进口某品牌)下青蒿素与杂质A、杂质B峰之间的分离度。结果均分离良好,对本品有关物质的检测没有显著影响。
2.11 3批样品测定取3批样品(批号:20141001、20141002、20141003),按“2.2”项下方法制备供试品溶液与对照溶液,按“2.1”项下色谱条件测定,结果见表1。
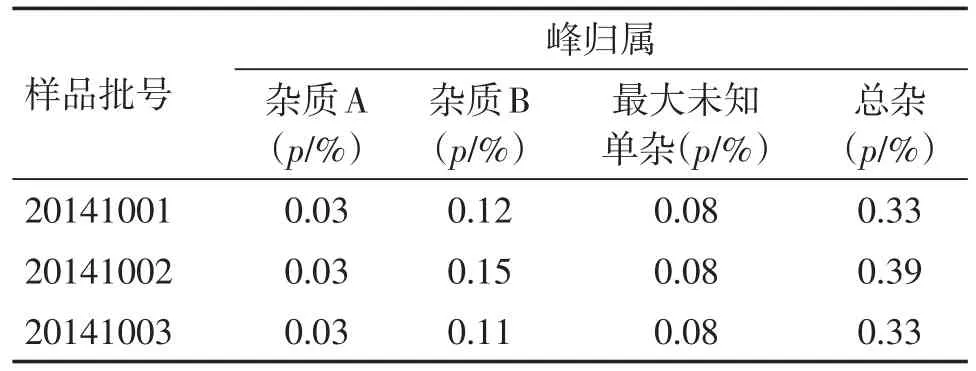
表1 3批样品有关物质测定结果Table 1 Determination results of related substances in 3 batches of samples
3 讨论
由原质量标准方法检测,杂质B未检出,反映原方法灵敏度差,但总杂含量2%,又与其青蒿素含量99%不一致,反映原方法检测结果准确性差。由此可见,原方法检测杂质B检测灵敏度低,检测结果杂质A、未知单杂和总杂虚高,与实际情况不符。
新修订质量标准方法采用乙腈-0.01%三氯乙酸系统的流动相,能将青蒿素峰、哌喹峰及其各杂质峰有效分开,检测结果准确度高。但三氯乙酸浓度不宜高,我们比较了0.01%、0.05%、0.1%3个浓度,发现酸浓度越高,对青蒿素和各杂质峰的峰面积影响较大,峰降解越严重。
从破坏试验看,各破坏条件下均降解产生未知杂质,但杂质A峰面积没有变化,较稳定。碱破坏条件和水溶液高温破坏条件下,杂质B峰面积增加明显,稳定性考察需重点关注。
耐用性试验显示,不同色谱柱对各峰的相对保留时间不一致,对杂质B的含量计算差异也大。相同品牌色谱柱对各峰的相对保留时间一致,杂质含量结果一致。建议使用该方法应注意流动相三氯乙酸的浓度配制要精准,以及要求使用固定品牌的色谱柱。
青蒿素原料中杂质B的含量较低,部分原料中杂质B几乎不能检出,参照《国际药典》中青蒿素原料中有关物质的方法,在系统适用性试验中规定了青蒿素峰与杂质A峰的分离度应大于4.0,并增加了相邻色谱峰间分离度应大于1.5。
本研究对精密度试验的样品分别用外标法和自身对照法计算并进行比较,杂质A用外标法计算含量为0.03%,sR为3.41%,用自身对照法计算含量为0.03%,sR为1.90%,合并两组数据含量为0.03%,sR为4.71%,表明两种方法计算结果一致。杂质B用外标法计算含量为0.31%,sR为3.03%,用自身对照法计算含量为0.39%,sR为4.15%,合并两组数据含量为0.35%,sR为12.70%,因杂质B校正因子为0.90(在0.9~1.1范围内),但以自身对照法计算时杂质B校正因子按1.00计算,又因用供试品溶液对照峰面积直接计算,故2种计算方法会有一定差异,但在允许范围内。参照《国际药典》青蒿素有关物质方法和2015年版《中国药典》(二部)青蒿素哌喹片中青蒿素有关物质方法,确定采用自身对照法计算,杂质A需加校正因子。
