HPLC法测定苯唑嗪胶囊中主成分及有关物质的含量
2019-09-10张英华高佳陈人萍刘卫王清张炜煜
张英华 高佳 陈人萍 刘卫 王清 张炜煜

摘 要 目的:建立测定苯唑嗪胶囊中主成分及有关物质含量的方法。方法:采用高效液相色谱法测定主成分含量,采用主成分自身对照法计算有关物质(已知杂质1、已知杂质2、总杂质)的含量。色谱柱为Inertsil ODS-2 C18,流动相为乙腈-水(55 ∶ 45,V/V),流速为1.0 mL/min,检测波长为223 nm,柱温为25 ℃,进样量为20 µL。结果:苯唑嗪主成分与杂质分离度良好,苯唑嗪检测质量浓度线性范围为20.04~60.12 µg/mL(r=1.000 0);精密度、稳定性(24 h)、重复性试验的RSD均≤0.5%(n=6);平均回收率为97.50%(RSD=0.36%,n=3);苯唑嗪的检测限和定量限分别为0.91、3.04 ng。在3批样品中,苯唑嗪、已知杂质1、已知杂质2、总杂质含量的平均值分别为106.68%、0.002 1%、0.044 0%、0.046 2%。结论:建立的苯唑嗪胶囊中主成分及有关物质的含量测定方法简便、专属性强、灵敏度高,结果准确,适用于苯唑嗪胶囊的质量控制。
关键词 苯唑嗪胶囊;高效液相色谱法;含量测定;有关物质
ABSTRACT OBJECTIVE: To establish a method for content determination of main components and its related substances in Phenzolzine capsules. METHODS: HPLC method was adopted for content determination of main components. The contents of related substance (known impurity 1, known impurity 2, total impurity) were calculated with principle component self-control method. The determination was performed on Inertsil ODS-2 C18 column with mobile phase consisted of acetonitrile-water (55 ∶ 45, V/V)at the flow rate of 1.0 mL/min. The detection wavelength was set at 223 nm, and column temperature was 25 ℃. The sample size was 20 µL. RESULTS: The main component phenzolzine and other impurity peaks were well separated. The liner range of phenzolzine was 20.04-60.12 µg/mL (r=1.000 0). RSDs of precision, stability (24 h) and reproducibility tests were all ≤0.5% (n=6). Average recovery was 97.50% (RSD=0.36%, n=3). The detection limit and quantification limit of phenzolzine were 0.91 ng and 3.04 ng. In 3 batches of samples, average value of phenzolzine, known impurity 1, known impurity 2 and total impurity were 106.68%, 0.002 1%, 0.044 0% and 0.046 2%, respectively. CONCLUSIONS: Established method is simple, specific, sensitive and accurate for content determination of main component and related substance in Phenzolzine capsules. It is suitable for quality control of Phenzolzine capsules.
KEYWORDS Phenzolzine capsules; HPLC; Content determination; Related substances
苯唑嗪(研發代号:Q808)化学名为6-(4-氯苯氧基)-四唑并[5,1-a]酞嗪,是一种新的抗癫痫药物,具有增强脑内核团自发性抑制性突触后电流(sIPSC)频率、增加γ-氨基丁酸(GABA)含量的作用,其保护指数(PI)优于卡马西平和卢非酰胺,在整体和离体癫痫模型中均具有较强的抗癫痫作用,与传统抗癫痫药物苯妥英钠、托吡酯相比,具有较强的抗惊厥作用且神经毒性较低[1]。以苯唑嗪为原料制成的苯唑嗪胶囊目前已获得临床试验批件,正在进行临床试验,在国内外尚未有关于该药质量标准的研究报道。为了保证用药安全、有效,笔者根据《化学药物稳定性研究的技术指导原则》[2]、《化学药物杂质研究的技术指导原则》[3]、《化学药物质量标准建立的规范化过程技术指导原则》[4]和2015年版《中国药典》(四部)[5]的相关要求,另参考相关文献[6-11],建立了以高效液相色谱(HPLC)法测定苯唑嗪胶囊中主成分含量及有关物质的方法,为该制剂的质量控制提供参考。
1 材料
1.1 仪器
LC-2010AHT型HPLC仪,包括二极管阵列检测器、色谱工作站(日本岛津公司);KQ-2200型超声波清洗仪(昆山市超声仪器有限公司,文中采用参数为功率:250 W,频率:40 kHz);BT125D型十万分之一电子天平(德国赛多利斯公司)。
1.2 药品与试剂
苯唑嗪对照品(批号:20140331,纯度:>99.9%)、5-氯四唑并[5,1-a]酞嗪对照品[已知杂质1[12](以下简称杂质1),批号:20140518,纯度:>99.9%]、6-(2,4-二氯苯氧基)-四唑并[5,1-a]酞嗪对照品[已知杂质2[12](以下简称杂质2),批号:20140518,纯度:>99.9%]均由延边大学药学院药物化学教研部制备;苯唑嗪胶囊(吉林省中医药科学院制剂室,批号:20140521、20140522、20140523,规格:0.15 g);乳糖(美国Foremost Farms公司);交联聚维酮(PVPP,国药集团化学试剂有限公司);微晶纤维素(湖州展望药业有限公司);聚乙烯己内酰胺-聚醋酸乙烯酯-聚乙二醇接枝共聚物(Soluplus®,德国巴斯夫有限公司);甲醇、乙腈均为色谱纯,水为纯净水,其余试剂均为分析纯。
2 方法与结果
2.1 色谱条件
色谱柱:Inertsil ODS-2 C18(250 mm×4.6 mm,5 µm);流动相:乙腈-水(55 ∶ 45,V/V);检测波长:223 nm;柱温:25 ℃;流速:1.0 mL/min;进样量:20 µL。
2.2 溶液的制备
2.2.1 对照品溶液 精密称取苯唑嗪对照品10.04 mg,置于50 mL量瓶中,加乙腈超声(功率:250 W,频率:40 kHz,下同)10 min;取出,放冷,稀释至刻度,摇匀,精密量取5 mL,置于25 mL量瓶中,加乙腈稀释至刻度,摇匀,制成每1 mL中含苯唑嗪40.16 μg的溶液,作为对照品溶液。
2.2.2 供试品溶液 精密称取苯唑嗪胶囊内容物适量(约相当于苯唑嗪10 mg),置于50 mL量瓶中,用乙腈超声10 min;取出,放冷,稀释至刻度,摇匀,精密量取5 mL,置于25 mL量瓶中,加乙腈稀释至刻度,摇匀,即得。
2.2.3 系统适用性溶液 精密称取杂质1、杂质2对照品各5 mg,置于10 mL量瓶中,用乙腈稀释至刻度,得系统适用性贮备液;精密称取苯唑嗪对照品10.06 mg,置于50 mL量瓶中;精密移取系统适用性贮备液1 mL,置于上述50 mL量瓶中,定容,摇匀,得系统适用性溶液。
2.2.4 空白辅料溶液 按样品处方精密称取除苯唑嗪外的空白辅料适量,再按“2.2.2”项下方法进行制备,即得空白辅料溶液。
2.3 系统适用性试验
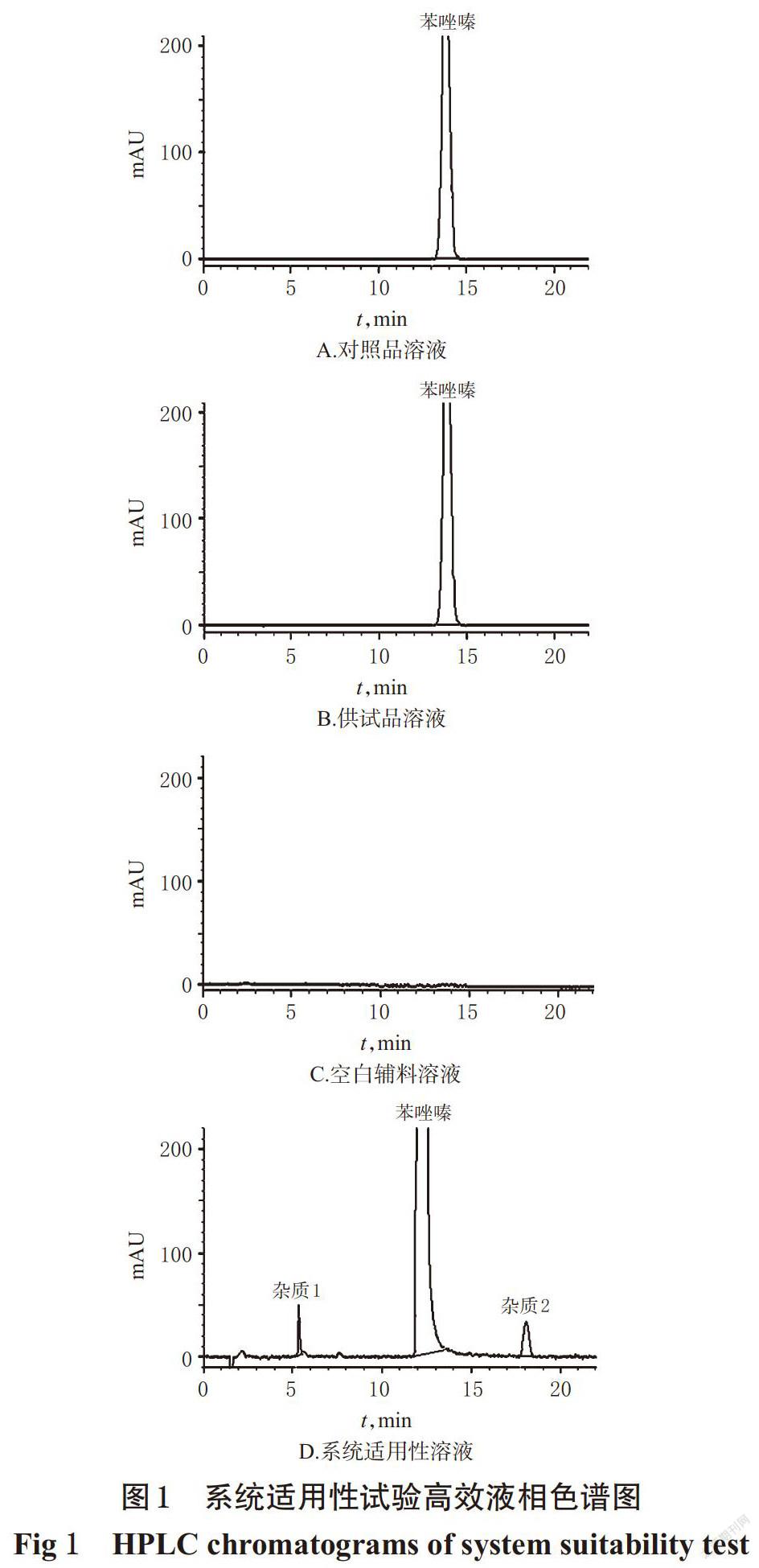
精密量取“2.2”项下对照品溶液、供试品溶液(批号:20140523)、系统适用性溶液及空白辅料溶液各20 μL,按“2.1”项下色谱条件进样分析。结果,杂质1、杂质2与苯唑嗪的分离度>5,其余各峰之间的分离度>1.5,主峰拖尾因子为1.14~1.16,理论板数以主成分峰计>5 000,详见图1。
2.4 方法专属性考察
取苯唑嗪胶囊10粒(批号:20140523)内容物研细,精密称取约5 mg,共5份,分别置于25 mL量瓶中:1份加入1 mol/L的盐酸溶液 5 mL,55 ℃水浴加热3 h,用1 mol/L的氢氧化钠溶液中和,调节至中性,加乙腈稀释至刻度,摇匀,作为酸破坏溶液;1份加入0.1 mol/L的氢氧化钠溶液 5 mL,静置3 h,用1 mol/L的盐酸溶液中和,调节至中性,加乙腈稀释至刻度,摇匀,作为碱破坏溶液;1份加入30%过氧化氢溶液5 mL,室温放置3 h,加乙腈稀释至刻度,摇匀,作为氧化破坏溶液;1份加入5 mL蒸馏水,70 ℃水浴加热3 h,加乙腈稀释至刻度,摇匀,作为高温破坏溶液;1份加入乙腈5 mL,使溶解,置于4 500 lx条件下照射18 h,加乙腈稀释至刻度,摇匀,作为光照破坏溶液。精密量取上述5种溶液及未破坏样品溶液各20 µL,分别注入色谱仪,按“2.1”项下色谱条件进样测定,记录其色谱图。结果表明,样品在酸、碱、氧化、高温和光照条件下均保持相对稳定,各破坏条件下产生的降解产物峰与主峰均能达到基线分离,且各峰间分离良好,详见图2。
2.5 线性关系考察
取苯唑嗪對照品10 mg,精密称定,置于50 mL量瓶中,加乙腈适量,超声溶解,用乙腈稀释至刻度,摇匀,作为贮备液。分别精密量取上述贮备液适量,用乙腈稀释制成质量浓度为20.04、24.05、32.06、40.08、48.10、60.12 µg/mL的系列质量浓度对照品溶液。分别精密量取20 µL,按“2.1”项下色谱条件进样测定,记录峰面积,以峰面积为纵坐标(y)、质量浓度(µg/mL)为横坐标(x)进行线性回归,得回归方程y=22 971 318.110x+95 894.154(r=1.000 0)。结果表明,苯唑嗪检测质量浓度线性范围为20.04~60.12 µg/mL。
2.6 检测限和定量限试验
分别取“2.5”项下苯唑嗪对照品贮备液适量,均采用逐步稀释法稀释至3.2 µg/mL,按“2.1”项下色谱条件进样测定,记录色谱图,以信噪比为3 ∶ 1计算[13]苯唑嗪的检测限,结果为0.91 ng;以信噪比为10 ∶ 1计算苯唑嗪的定量限,结果为3.04 ng。
2.7 精密度试验
精密量取“2.2.1”项下的苯唑嗪对照品溶液适量,按“2.1”项下色谱条件重复进样6次,记录峰面积,结果苯唑嗪峰面积的RSD为0.46%(n=6),表明仪器精密度良好。
2.8 稳定性试验
取“2.2.2”项下供试品溶液(批号:20140523)适量,分别在室温下放置0、2、4、8、12、24 h后,按“2.1”项下色谱条件进样测定,计算得到苯唑嗪峰面积的RSD为0.33%(n=6),表明供试品溶液在室温下放置24 h内稳定性良好。
2.9 重复性试验
取苯唑嗪胶囊样品(批号:20140523)适量,平行6份,分别按“2.2.2”项下方法制备供试品溶液,按“2.1”项下色谱条件进样测定,得平均含量为99.98%,RSD为0.18%(n=6),表明本方法重复性良好。
2.10 加样回收率试验
按样品处方精密称取空白辅料适量,共9份。精密称取苯唑嗪对照品适量,置于50 mL量瓶中,加乙腈适量,超声溶解,再用乙腈稀释至刻度,作为对照品溶液,分别取低、中、高不同体积的溶液各3份(含苯唑嗪的量分别约为400、500、600 µg),加入至9份空白辅料中,用乙腈稀释并定量,取溶液按“2.1”项下色谱条件测定并计算回收率,结果见表1。
2.11 耐用性试验
取苯唑嗪胶囊样品(批号:20140523)适量,按“2.2.2”项下方法处理制备供试品溶液,在选定流动相系统下,对不同色谱柱(色谱柱1:Agilent TC-C18,250 mm×4.6 mm,5 µm;色谱柱2:Diamonsil-C18,250 mm×4.6 mm,5 µm;色谱柱3:Inertsil ODS-2 C18,250 mm×4.6 mm,5 µm)进行比较,结果发现采用不同色谱柱进行分析时,主峰与杂质峰均能得到良好的分离,但以采用Inertsil ODS-2 C18色谱柱时峰形最佳。
在其他色谱条件不变情况下,考察流动相不同体积比[乙腈-水(52 ∶ 48、58 ∶ 42、55 ∶ 45,V/V)]、不同柱温(25、30、35 ℃)、不同流速(0.8、1.0、1.2 mL/min)及不同流动相pH(3、5、7)对含量测定结果的影响,结果,4种变化条件下(各平行2次)苯唑嗪峰面积的RSD分别为0.22%(n=6)、0.19%(n=6)、0.20%(n=6)、0.26%(n=6),表明本方法耐用性良好。
2.12 样品含量及有关物质的检查
2.12.1 样品含量测定 精密称取苯唑嗪对照品和苯唑嗪胶囊3批样品适量,按“2.2”项下方法制备对照品溶液和供试品溶液,并按“2.1”项下色谱条件进样测定,记录峰面积,采用外标法以峰面积计算含量[14],结果见表2。
2.12.2 样品有关物质检查 取苯唑嗪胶囊3批样品适量,精密称定,分别加乙腈稀释制成0.32 mg/mL的溶液,作为有关物质供试品溶液;精密量取上述有关物质供试品溶液1 mL,置于100 mL量瓶中,加乙腈稀释至刻度,摇匀,作为有关物质对照溶液。量取对照溶液20 µL,注入HPLC仪,调节检测灵敏度,使主成分色谱峰高约为满量程的20%;再精密量取有关物质供试品溶液与有关物质有关物质对照溶液各20 µL,分别注入色谱仪,记录色谱图至主成分保留时间的2倍。在有关物质供试品溶液的色谱图中如出现除空白辅料外的杂质峰,按主成分自身对照法计算有关物质的含量(单个杂质不得过0.1%,总杂质不得过0.3%)[15-17],结果见表2。
由表2数据计算可知,在3批样品中,苯唑嗪、杂质1、杂质2、总杂质含量的平均值分别为106.68%、0.002 1%、0.044 0%、0.046 2%。
3 讨论
苯唑嗪胶囊是针对癫痫治疗研发的化学药品1.1类新药[18],目前,相关研究已对苯唑嗪原料药中的杂质进行了分离鉴定及初步的测定[12],但并未对其进行系统的质量检查方法研究,特别是对苯唑嗪胶囊,目前国内外尚无相关研究的文献报道。本试验采用HPLC法,对苯唑嗪胶囊主成分的含量及有关物质进行了深入研究,以更好、更全面地控制该制剂的质量。
在色谱条件优化过程中,笔者主要考察了流动相组成以及检测波长对分离效果的影响。在流动相组成中,分别比较了甲醇-水和乙腈-水为流动相的分离效果,结果表明,选择乙腈-水(55 ∶ 45,V/V)为流动相时,主成分与各杂质间的分离效果良好(分离度>2),保留时间(杂质1:5.355 min;主成分:12.224 min;杂质2:18.088 min)合适,且主成分与杂质均能满足分析要求。采用二极管阵列检测器对检测波长进行筛选,发现各成分在223 nm波长处有最大吸收,且灵敏度较高。故选择223 nm作为本研究的检测波长。
综上所述,本研究建立的苯唑嗪胶囊中主成分及有关物质的含量测定方法简便、专属性强、灵敏度高,结果准确,适用于苯唑嗪胶囊的质量控制。
参考文献
[ 1 ] SUN XY,WEI CX,DENG XQ,et al. Evaluation of the anticonvulsant activity of 6-(4-chlorophenyoxy)-tetrazolo[5,1-a]phthalazine in various experimental seizure models in mice[J]. Pharmacol Rep,2010,62(2):273-277.
[ 2 ] 国家食品药品监督管理局.化学药物稳定性研究的技术指导原则[S]. 2005-03-18.
[ 3 ] 国家食品药品监督管理局.化学药物杂质研究的技术指导原则[S]. 2005-03-18.
[ 4 ] 国家食品药品监督管理局.化学药物质量标准建立的规范化过程技术指导原则[S]. 2005-03-18.
[ 5 ] 国家药典委员会.中华人民共和国药典:四部[S]. 2015年版.北京:中国医药科技出版社,2015:375-378.
[ 6 ] 李维宏,罗静,王高升.草铵膦原药的高效液相色谱分析[J].浙江化工,2009,40(11):24-26.
[ 7 ] 陳玲,徐洪.高效液相色谱法测定丙磺舒原料药含量的研究[J].贵阳中医学院学报,2010,32(5):80-83.
[ 8 ] 高微,邵海云,张吉娟,等. HPLC法测定氢溴酸山莨菪碱片的含量及含量均匀度[J].中国药房,2015,26(9):1284- 1285.
[ 9 ] 谢沐风.如何建立高效液相色谱法测定有关物质的方法[J].中国医药工业杂志,2007,38(1):45-48.
[10] 赵慧玲.浅谈药品注册中HPLC法测定原料药有关物质的问题[J].药物分析杂志,2007,27(6):947-948.
[11] 王彪,张翠莲,梅丹.不同厂家克拉霉素制剂含量、有关物质及溶出度的对比研究[J].中国药学杂志,2014,49(21):1933-1938.
[12] 李龙,刘冲,全哲山,等.抗癫痫一类新药Q808有关物质的结构确定及含量测定[J].延边大学医学学报,2016,39(2):103-106.
[13] 鲁静,付凌燕,王旭.质量分析方法验证中检出限和定量限测定方法探讨[J].中国药品标准,2012,13(1):33-35.
[14] 粟贵,刘雁鸣,龙海燕,等. HPLC法测定并比较药用级与非药用级苯甲酸钠的含量[J].中国药房,2016,27(18):2566-2569.
[15] 刘士敏,白若岑,肇鑫宇,等.脱氧核苷酸钠制剂的有关物质研究[J].沈阳药科大学学报,2015,32(6):457-462.
[16] 李霞,吉红梅,柳小秦,等. HPLC法测定盐酸硫利达嗪片的有关物质及含量[J].药物分析杂志,2015,35(4):732- 736.
[17] 黄姗,张梦泽. HPLC法测定维格列汀片中的有关物质[J].中国药房,2017,28(15):2138-2141.
[18] 张凤,姜昊澄,陈香儒,等. 6-(4-氯苯氧基)-四唑并[5,1-a]酞嗪对小鼠的抗抑郁样作用[J].中国药理学与毒理学杂志,2013,27(5):783-788.
(收稿日期:2019-04-01 修回日期:2019-05-27)
(编辑:刘 萍)
