两种基于酰腙配体的镍配合物:水热合成、结构、抗癌活性与量化计算
2019-09-09陈延民解庆范
陈延民 解庆范
(泉州师范学院化工与材料学院,泉州 362000)
酰腙类Schiff碱的热稳定性好,不易水解,与金属离子具有很强的配位能力,在催化剂、功能材料和荧光分析等方面有广泛应用,而且许多金属的酰腙配合物具有良好的抗癌、抑菌和抗病毒等生物活性,因此长期以来备受研究者的关注[1-8]。由酰肼与水杨醛衍生物缩合的产物是一类[ONO]三齿配体,与金属可以形成稳定的螯合物,利用咪唑、吡啶和4,4′-联吡啶等第二配体的辅助作用,可以设计合成具有大共轭平面的配合物,而这种大平面往往有利于配合物与DNA的相互作用。镍是生物体中必需的微量元素,能促进体内铁的吸收、红细胞的增长和氨基酶的合成,而且可能是DNA和RNA的一种结构稳定剂[9-11],有些镍配合物还表现出良好的抗癌活性[12-15]。本文采用水热法合成了两种酰腙的镍配合物[Ni(Lss)(Py)](1)和[Ni2(Lrr)2(4,4′-bipy)](2),经X射线单晶衍射方法确定了它们的晶体结构,采用MTT法测试了配合物对白血病细胞HL-60体外抑制活性。同时采用密度泛涵理论(DFT)对配合物2分子的自然电荷布居和键级进行了分析。
1 实验部分
1.1 仪器与药品
所用仪器有德国Elmentar公司 Vario EL型元素分析仪,美国Nicolet公司is10型FT-IR红外光谱仪,上海美普达UV-1800PC型紫外-可见分光光度计,德国Bruker公司Smart Apex CCD单晶衍射仪。5-硝基水杨醛缩噻吩-2-甲酰腙(H2Lss,按文献[16]方法自制),5-溴水杨醛缩噻吩-2-甲酰腙(H2Lrr,按文献[17]方法自制),其他均为市售的分析纯试剂。
1.2 配合物的合成
将 0.1 mmol H2Lss,0.1 mmol乙酸镍和 20 μL 吡啶(约0.25 mmol)以及2 mL蒸馏水和5 mL甲醇,置于20 mL的内衬聚四氟乙烯的不锈钢自动升压反应釜中,在140℃下反应2 d,得到适于X射线单晶衍射分析的1的黄色晶体。对C17H12N4NiO4S元素分析的实测值(计算 值,%):C 47.74(47.81);H 2.76(2.83);N 13.15(13.11)。 IR(cm-1):1 602(s),1 553(m),1 512(w),1 485(w),1 426(w),1 332(s),1 313(vs),1 219(m),927(w),828(w),768(w),725(s),695(m)。
将 0.2 mmol H2Lrr,0.2 mmol乙酸镍和 0.1 mmol 4,4′-联吡啶以及3 mL蒸馏水和7 mL甲醇,同样在140℃下水热反应2 d,得到2的黄色晶体。对C34H22Br2N6Ni2O4S2元素分析的实测值 (计算值,%):C 44.31(44.39);H 2.52(2.41);N 9.22(9.14)。 IR(cm-1):1 622(m),1 594(s),1 560(m),1 457(vs),1 419(m),1 372(s),1 300(s),1 174(s),948(vw),845(w),811(m),722(s),694(w)。
1.3 晶体结构测试
选取1和2的单晶置于Smart Apex CCD单晶衍射仪上,用经石墨单色器单色化的Mo Kα射线(λ=0.071 073 nm) 分别在 3.14°<θ<27.54°和 3.38°<θ<25.10°范围内以φ~ω扫描方式分别收集到65 940和25 599个衍射点,其中I>2σ(I)的可观察点分别为2 242和1 521个。晶体结构用SHELXTL和SHELXL-97程序包[18-19]由直接法解出,全部强度数据均经Lp因子校正,并进行了经验吸收校正,对全部非氢原子坐标及其各向异性热参数进行全矩阵最小二乘法修正,氢原子由理论加氢法得到。晶体学数据详见表1,主要键长和键角列于表2。
CCDC:1041669,1;1046102,2。

表1 配合物1和2的主要晶体数据Table 1 Crystal data of complexes 1 and 2

续表1
2 结果与讨论
2.1 晶体结构描述
配合物1是一种单核配合物,分子结构见图1。它由1个酰腙配体Lss2-、1个吡啶配体和1个Ni2+离子组成,其中,中心离子Ni(Ⅱ)为4配位,处于平面四边形的配位环境,4个配原子分别来自烯醇化的酰腙配体的羰基氧原子O、去质子化的羟基氧原子O、亚胺基的氮原子N和吡啶的氮原子N。Ni-O键长为0.184 5(2)和 0.182 3(2)nm,Ni-N键长为 0.183 2(3)和 0.194 5(3)nm。由于羰基烯醇式配位,所以O(1)-C(10)键长(0.130 0 nm)与酚羟基 O(2)-C(13)键长(0.131 1 nm)相近,明显比类似的酰腙配体[20]的羰基C=O双键键长(0.122 nm)长得多。配原子N1、N3、O1和O2与中心离子 Ni2+几乎完全共平面,键角为 83.51(11)°~94.84(11)°和 174.92(12)°~177.71(10)°,O1-N1-O2-N3 扭转角为-1.71°。配合物整个分子也呈现较高的共平面性,噻吩环与苯环二面角为9.2°,吡啶与酰腙配体共轭面的二面角为10.5°;扭转角O1-C10-N2-N3和N3-C11-C12-C13分别为-1.52°和1.37°,说明配合物1整个分子的共轭程度较高。
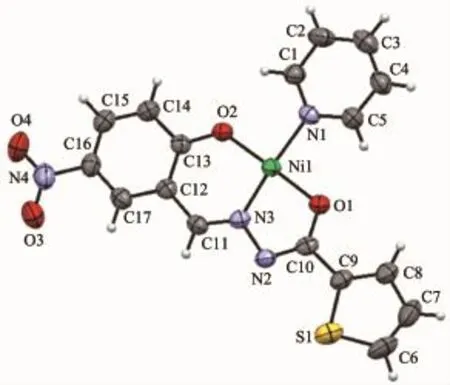
图1 配合物1的分子结构图(椭球率30%)Fig.1 Molecular structure of 1 with 30%probability ellipsoids
配合物2的分子结构见图2。它是由2个酰腙配体 Lrr2-、1 个 4,4′-联吡啶和 2 个中心离子 Ni(Ⅱ)组成的具有对称中心的双核配合物。与配合物1相似,烯醇化的酰腙配体去质子化,提供2个氧原子和1个氮原子与吡啶环的氮原子构成平面四边形的配位环境,Ni-O键长为0.181 9(4)和0.184 3(4)nm,Ni-N键长为0.183 2(3)和 0.193 8(4)nm;键角为 83.81(19)°~95.7(2)°和 173.7(2)°~177.64(17)°。 酰腙配体 Lrr2-的所有原子与NI(Ⅱ)共平面程度高,噻吩环与苯环二面角为5.4°,O1-Ni1-N2-N1和N2-N1-C5-O1扭转角分别为 2.2(3)°和1.5(7)°,O1-N3-O2-N2 扭转角为 3.8(3)°。4,4′-联吡啶的所有原子完全共平面,它与酰腙配体共轭平面的二面角为 16.1°。
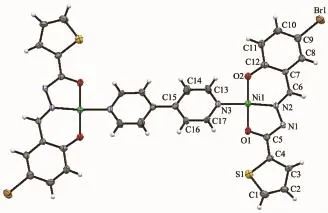
图2 配合物2的分子结构图(椭球率30%)Fig.2 Molecular structure of 2 with 30%probability ellipsoids
在1的晶体中,每个配合物分子的噻吩环的C-H和吡啶环的C-H与相邻配合物分子的硝基氧原子O之间形成氢键(表3),从而将整个配合物扩展为二维超分子体系(图3),而这些氢键对于稳定晶体结构起着重要作用。而2则通过芳环堆积作用形成层状超分子体系。
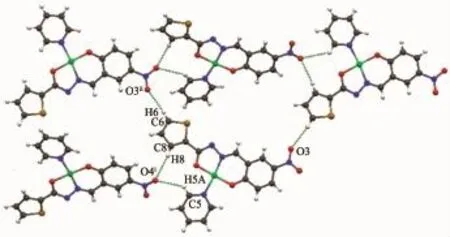
图3 配合物1中的分子间氢键Fig.3 Intermolecular C-H…O hydrogen bonds in the crystal of 1
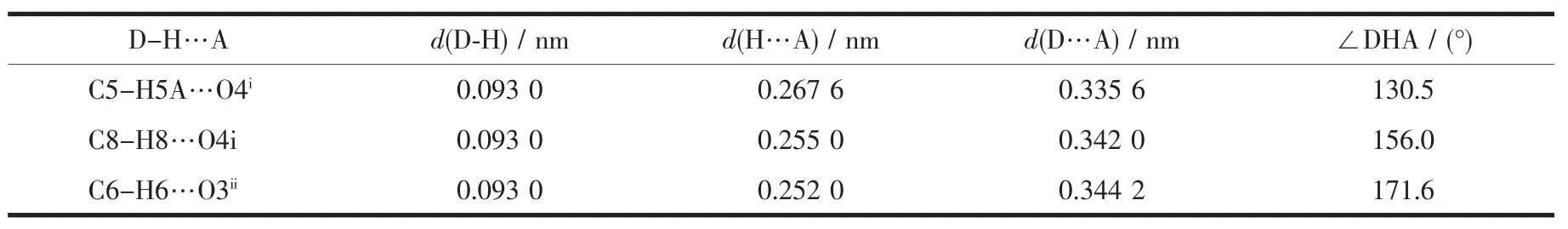
表3 配合物1中的氢键键长(nm)和键角(°)Table 3 Hydrogen bonds in the crystal of 1
2.2 电子光谱特征
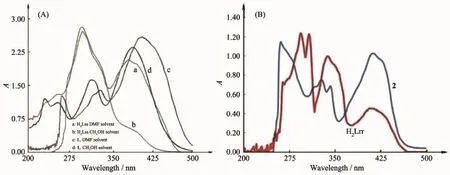
图4 配体和配合物的紫外-可见光谱Fig.4 UV-Vis spectra of ligands and complexes
配体和配合物溶液(21.8 μmol·L-1)的电子光谱见图4。由图4(A)可见,在DMF溶剂中H2Lss有2个很宽的强吸收带,其中297 nm可指认为共轭体系的π-π*电子跃迁即K带和n-π*电子跃迁即R带叠加的结果,395 nm处则归属于配体内部的电荷转移跃迁(ILCT);而形成配合物1后,K带红移到328 nm,这是因为酰腙以烯醇式配位,共轭体系增大的结果,ILCT则红移至402 nm,在260 nm处出现的新吸收带可能来自第二配体吡啶环的π-π*电子跃迁即B带。溶剂对电子光谱的吸收特征有很大的影响,由于甲醇与配合物可形成氢键,有可能改变分子轨道的能级,从而使1的B带、K带和ILCT分别蓝移至230、312和386 nm;同时在252 nm处可以观察到R带。H2Lrr在265 nm处的肩峰归属芳环的B带,294、306和339 nm可能来自不同共轭片段的π-π*电子跃迁 (即K带吸收峰),405 nm 则来自 ILCT; 由于配合物 2 中 4,4′-联吡啶参与配位,所以在图4(B)中261 nm处明显可观察到2的B带;而酰腙以烯醇式配位导致K带红移至328~342 nm,电荷转移跃迁红移了8 nm至413 nm。
2.3 量化计算
用Gaussian 09程序包[21],应用密度泛函理论(DFT)[22],在 B3LYP 水平上对 C,H,O,N,S 原子选用 6-31G(d)基组,Ni原子选用lanl2dz基组,计算了配合物2的分子的碎片对前线分子轨道的贡献、Wiberg键级和自然电荷布居(表4~6)。计算中收敛精度采用程序的默认值。
配合物2的最高占据分子轨道的能量EHOMO为-5.368 eV,最低空轨道的能量ELUMO为-2.857 eV,能量较低,ELUMO与EHOMO的差值(2.511 eV)较大,说明配合物的稳定性较好。最高占据轨道(HOMO)电子云主要集中在酰腙C8N2O2Br(76.5%)片段和噻吩C4S(15.4%)片段,最低空轨道(LUMO)电子云主要集中在吡啶配体C5N(95.7%),表明电子由HOMO向LUMO跃迁时主要发生酰腙配体向4,4′-联吡啶配体的配体间电荷转移跃迁(LLCT),最大吸收波长的计算值为493 nm,实验值为413 nm。而LUMO+1电子云的分布是C8N2O2Br片段68.9%、C4S片段21.5%和C5N片段4.2%,可见电子由HOMO向LUMO+1跃迁时主要发生的是酰腙共轭体系的π-π*电子跃迁(即所谓的K带),最大吸收波长的计算值为351 nm,实验值为328~342 nm。
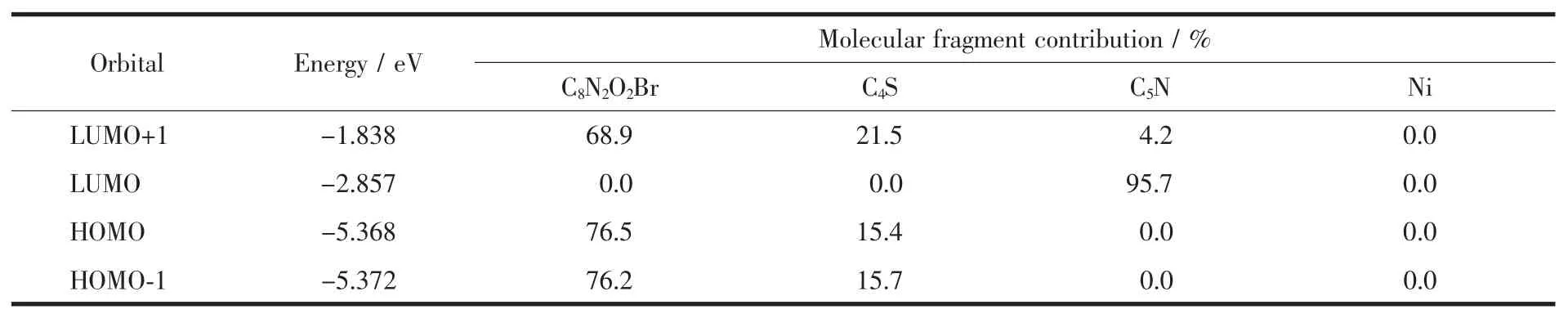
表4 配合物2的前线分子轨道能量和分子碎片对该分子轨道贡献/%Table 4 Frontier molecular orbital energy and molecular fragment contribution to molecular orbitals of 2
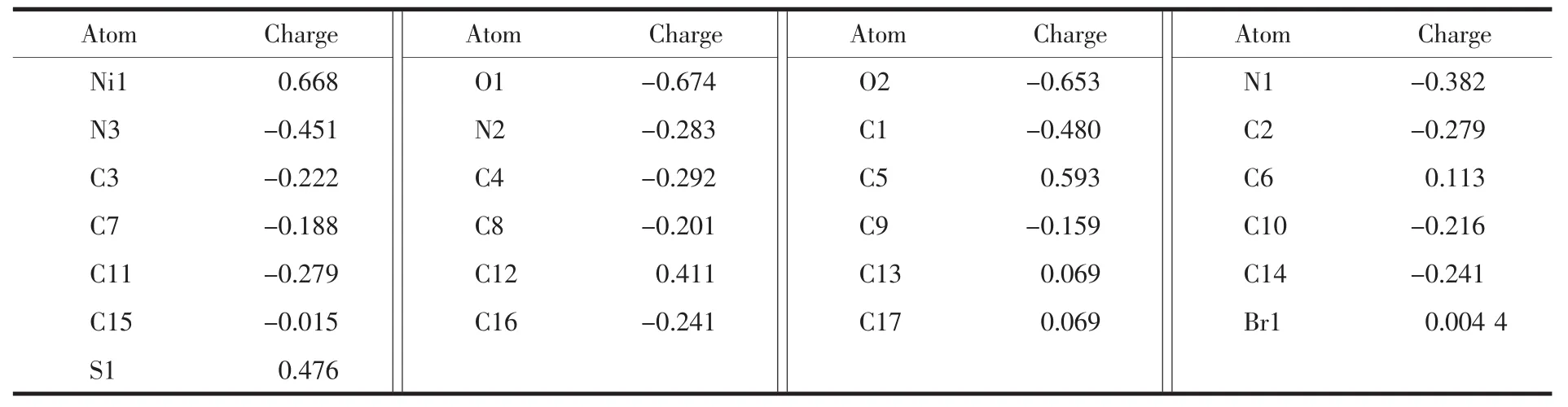
表5 配合物2的NBO电荷分布Table 5 NBO charges populations of 2
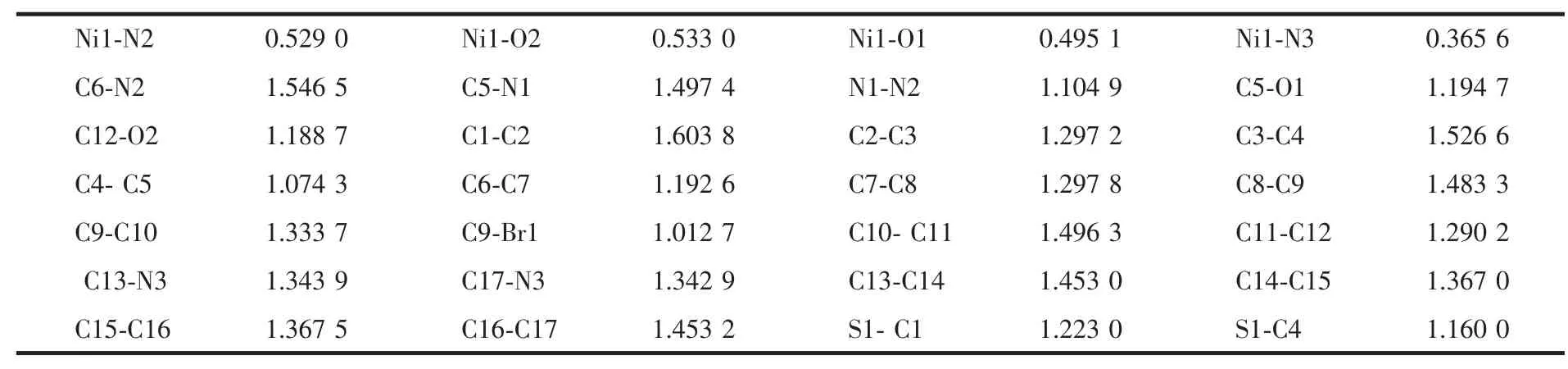
表6 配合物2的Wiberg键级Table 6 Wiberg bond order of 2
镍在配合物2中的化合价为+2,由表5可见,Ni1、O1、O2、N2 和 N3 的净电荷分别为 0.668、-0.674、-0.653、-0.283和-0.451,说明配原子的部分负电荷转向了中心离子,这从理论上证明了配位键的存在。电荷转移的结果使得与配原子相连的C5、C6、C12、C13和C17成为正电荷较为集中的原子。从表6可知,C5-N1和C6-N2的Wiberg键级分别为1.497和1.546 5,属典型的双键;C5-O1的键级为1.194 7,明显小于1.500,证明羰基以烯醇式配位。 Ni1-O1、Ni1-O2、Ni1-N2和Ni1-N3的键级分别为0.495 1、0.533 0、0.529 0和0.365 6,说明配位能力大小顺序为:酚羟基氧O2>亚胺基N2>羰基O1>吡啶基N3;同时说明Ni1-N1在热分解时可能优先断裂,成为热解引发键。
2.4 体外抗癌活性
在无菌条件下,分别取对数生长期的人体急性早幼粒白血病细胞HL-60,用2.5 g·L-1胰酶消化,调整细胞密度为2.5×104mL-1,培养24 h,用全自动酶标仪在570 nm处测定它的吸光度值并计算细胞的增殖抑制率,根据线性回归方程求出化合物的半数抑制浓度IC50。结果表明,配体H2Lss和H2Lrr的抗癌活性较弱,而配合物1和2对白血病细胞HL-60有较强的增殖抑制作用,且与浓度呈现正相关性 (图5)。1和2对HL-60半数抑制浓度 IC50分别为 0.476和 3.85 μg·mL-1(即 1.11 和 4.19 μmol·L-1)。根据 Shier[23]的建议,当IC50小于5 μg·mL-1时化合物的抑制活性为强效。从而进一步证明,大平面结构的过渡金属配合物具有潜在的生物活性。H2Lss和H2Lrr的结构相似,但1的抗癌活性明显高于2,可能与酰腙的取代基有关,因为硝基是强吸电子共轭基团,而溴是供电子共轭基团,吸电子效应有助于提高药物的抗癌活性[24]。
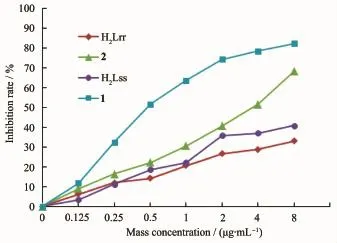
图5 化合物对癌细胞HL-60的抑制作用Fig.5 Inhibition effects of compounds on cell HL-60
