HPLC法测定盐酸左旋咪唑片的含量研究
2019-09-06周芷锦林仙军王彬陈晓林
周芷锦,林仙军,王彬,陈晓林
(浙江省兽药饲料监察所,杭州311101)
左旋咪唑是一种广谱驱虫药,广泛应用于兽医临床治疗畜禽胃肠道线虫病、肺丝虫病和猪肾虫病当中。除作为驱虫药之外,盐酸左旋咪唑还具有免疫调节作用,是一种免疫增强剂,能提高巨噬细胞的吞噬能力,增强机体的体液免疫功能和细胞免疫功能[1]。研究发现,盐酸左旋咪唑可显著提高脾脏指数和法氏囊指数,对雏鸡有显著的增重效果,在一定程度上提高生长性能[2]。目前兽医使用中常见的剂型包括盐酸左旋咪唑片、盐酸左旋咪唑注射液。目前盐酸左旋咪唑片在《中国兽药典(2015年版)》一部[3],《美国药典》(USP40-NF35)[4]中均有收入。《中国兽药典(2015年版)》一部[3]中,含量测定标准采用氯仿提取,高氯酸滴定的方法。该方法主要存在氯仿毒性较大,且过程较为繁琐,容易出现误差较大等问题。《美国药典》[4]采用高效液相色谱法,但对实验室仪器设备有较高的要求。基于文献参考[4-6],本着优化简化实验条件和方法的目的,本文探讨用高效液相色谱法测定盐酸左旋咪唑片中盐酸左旋咪唑含量的方法研究。
1 仪器与材料
1.1 仪器 高效液相色谱仪,Angilent 1260,配Angilent DAD检测器;METTLER TOLEDO XS205电子天平;KQ-500E型超声波清洗器(昆山市超声仪器有限公司);METTLER TOLEDO 320酸度计。
1.2试剂 乙腈、甲醇均为色谱纯,德国默克公司;磷酸二氢钾、磷酸、三乙胺均为分析纯;水为超纯水。盐酸左旋咪唑对照品来源为中国食品药品检定研究院,批号100167-201203,含量99.9%。
1.3 样品 实验样品由杭州新港动物药业有限公司等四家单位提供,4个批次25mg规格的样品批号分别为:20190101,180529,190122,20180703;2个批次50 mg规格的样品批号分别为:170901,190301。
2 方法与结果
2.1 溶液的配制
2.1.1 对照品溶液配制 取盐酸左旋咪唑对照品约10 mg,精密称定,置200 mL容量瓶中,加入适量流动相溶解,并用流动相稀释至刻度,摇匀。
2.1.2 供试品溶液配制 取本品20片,精密称定,研细,精密称取适量(约相当于盐酸左旋咪唑50 mg),置100 mL容量瓶中,加入流动相适量,超声10 min溶解,用流动相稀释至刻度,摇匀;过滤,精密量取续滤液5 mL,置50 mL容量瓶中,用流动相定容,摇匀。
2.2 色谱条件及系统适应性试验 采用十八烷基硅烷键合硅胶色谱柱(Agilent Eclipse XDB-C18,4.6 mm×250 mm,5 μm),流动相为0.05 mol/L磷酸二氢钾溶液(三乙胺调节pH值至7.0)-乙腈(80∶20,v/v);进样量:20 μL;柱温:30 ℃;流速:1.0 mL/min;用二极管阵列检测器在190~400 nm范围内进行扫描,记录214 nm波长处的色谱图。盐酸左旋咪唑对照品光谱图(图1),盐酸左旋咪唑对照品溶液色谱图(图2),供试品溶液色谱图(图3)。
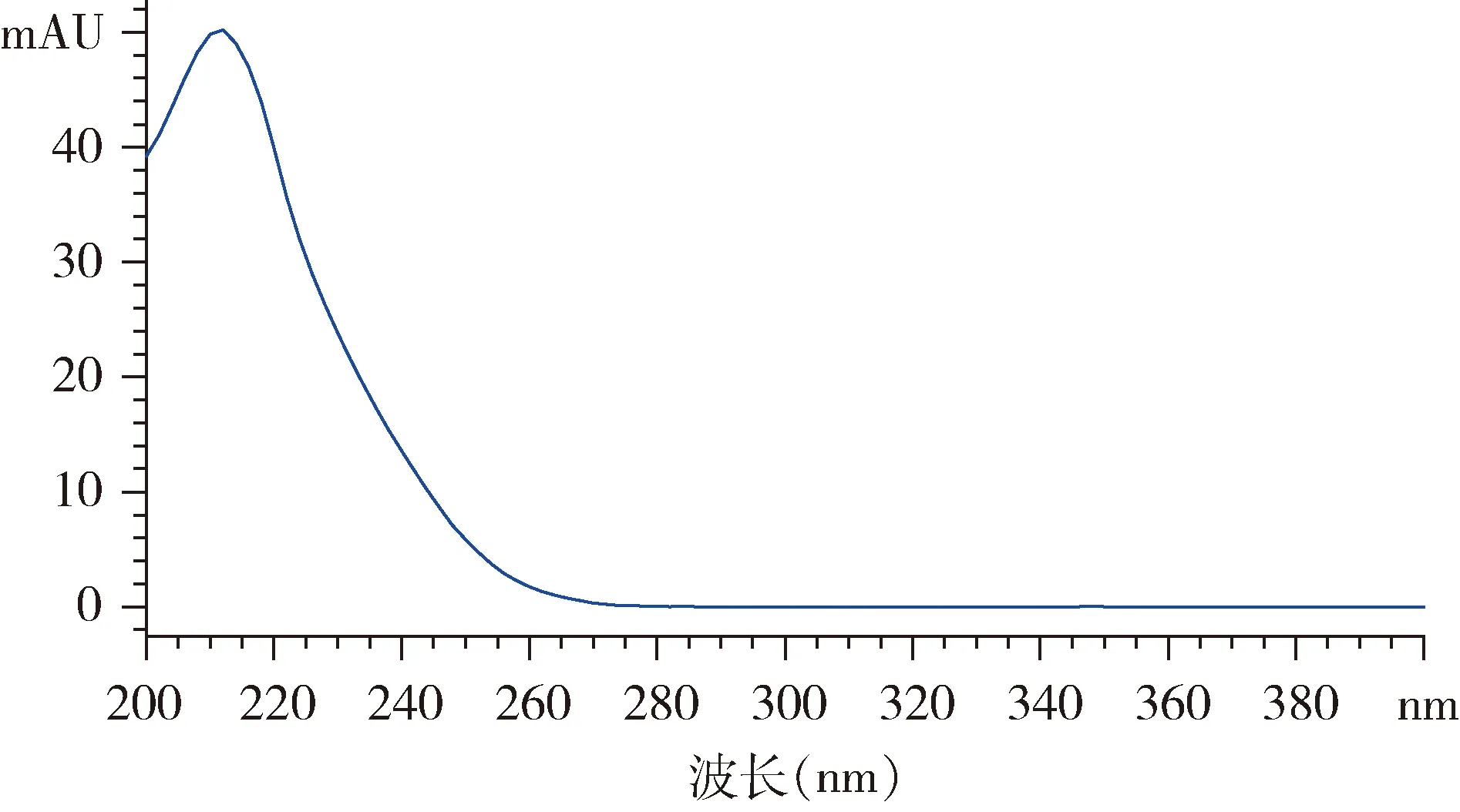
图1 盐酸左旋咪唑对照品溶液光谱图Fig 1 Spectrogram of Levamisole Hydrochloridereference substance
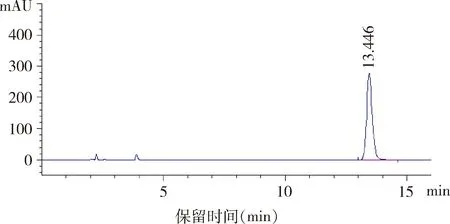
图2 盐酸左旋咪唑对照品溶液色谱图Fig 2 Solution chromatogram of LevamisoleHydrochloride reference substance
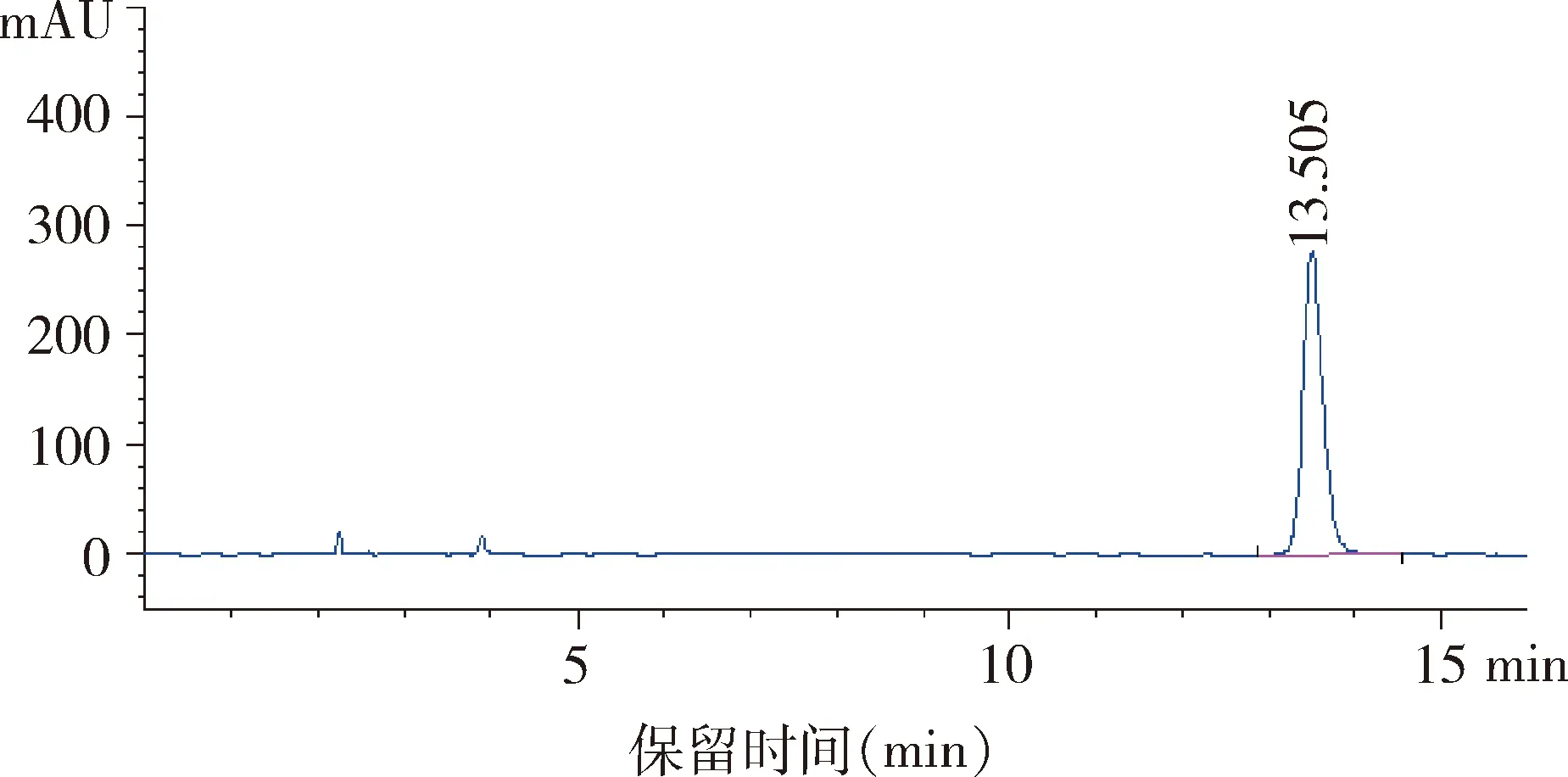
图3 供试品溶液色谱图Fig 3 Solution chromatogram of the sample
2.3 线性关系考察 取盐酸左旋咪唑对照品,精密称定,加流动相制成100 μg/mL的溶液,并配制成1、2.5、5、10、25、50、100 μg/mL等一系列浓度的对照品溶液。分别精密吸取20 μL,注入色谱仪,测定。以对照品浓度(X)为横坐标,峰面积值(Y)为纵坐标进行线性回归,绘制标准曲线。在1.024~102.398 μg/mL范围内线性关系良好,线性回归方程为Y=89.715X+14.765,线性相关系数为1.0000。
2.4 回收率试验 以批号190301的样品作为本底样品,精密称取9份,每三份为一组,称取量为应称取样品量的一半(约相当于盐酸左旋咪唑25 mg),置100 mL容量瓶中;另精密称取盐酸左旋咪唑对照品,高、中、低浓度对照品加入量按照应称取量的80 %、100 %、120%的比例加入上述的三组100 mL容量瓶中,并按照2.1.2的方法配制回收率试验用溶液。按照2.2色谱条件,精密量取20 μL,注入色谱仪中,记录色谱图。计算回收率,回收率=(实测量/加入量)×100 %,实测添加量=测得总量-本底盐酸左旋咪唑量。回收率结果如表1所示,平均回收率为97.4 %、99.7 %、99.3 %,说明检测方法的准确度良好。
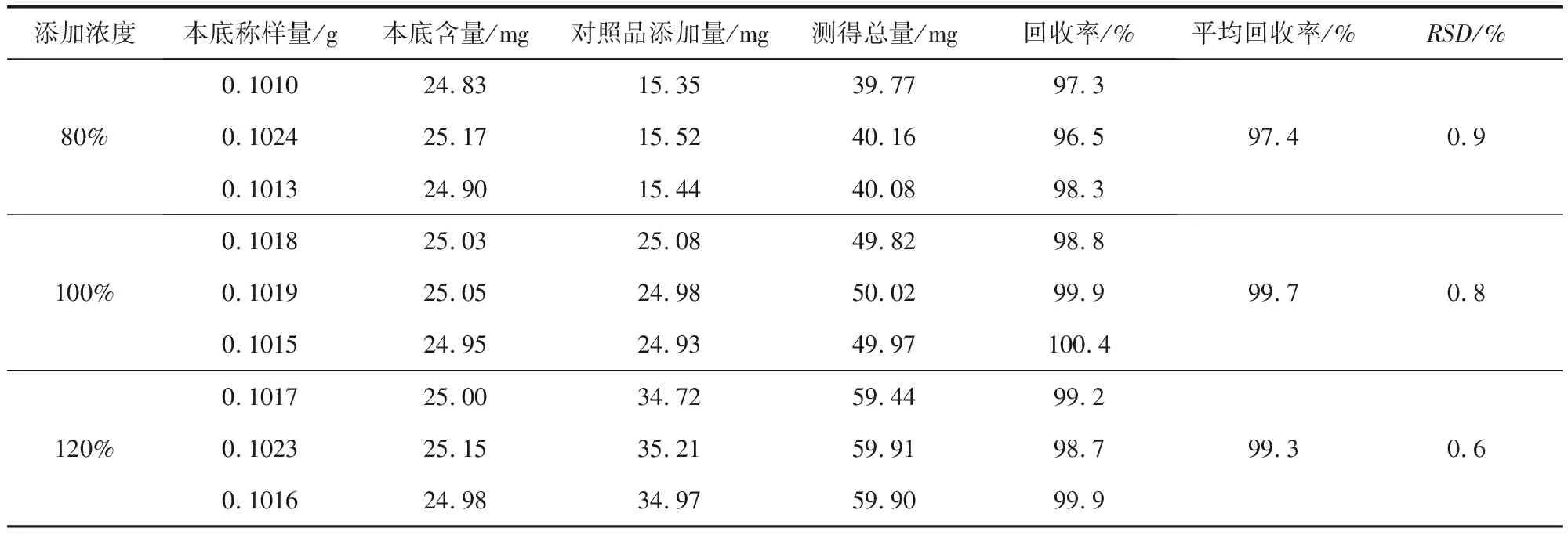
表1 回收率结果表Tab 1 Results of recovery tate
2.5 精密度 取回收率试验的9份样品测定,RSD值按照高、中、低浓度三组分别为0.9%,0.8%,0.6%,说明精密度良好。
2.6 稳定性试验 取重复性试验的供试品溶液,在0,1.5,3,6,12,16 h时间点进样,测得器峰面积分别为4483.87744,4474.66260,4492.76318,4482.00439,4516.70264,4501.69922,其检测结果的RSD为0.3%,说明在16 h内供试品溶液基本稳定。
2.7 耐用性试验 采用三台不同的液相和三根不同品牌的色谱柱分三组对批号为20190101的样品进行含量测定。A组为Agilent 1260 DAD液相色谱仪,配Agilent C18(4.6 mm×250 mm,5 μm)色谱柱;B组为Agilent 1260 VWD液相色谱仪,配Waters C18(4.6 mm×250 mm,5 μm)色谱柱;C组为Waters 2695-2487液相色谱仪,配Agilent C18(4.6 mm×150 mm ,5 μm)液相色谱柱。样品配制方法如2.1.2,色谱条件见2.2,含量测定结果如表2所示,同一样品三组含量测定结果分别为100.5%,101.3%,101.2%,RSD值为0.5%,说明该检测方法对液相和色谱柱品牌无特殊要求,耐用性良好。
2.8 实际样品含量测定 取1.3中的所有样品,每个样品平行3次,依法测定其含量,结果见表3。

表2 不同液相与不同品牌色谱柱的含量测定比较Tab 2 Comparison of content determination of different HPLC and different brand columns
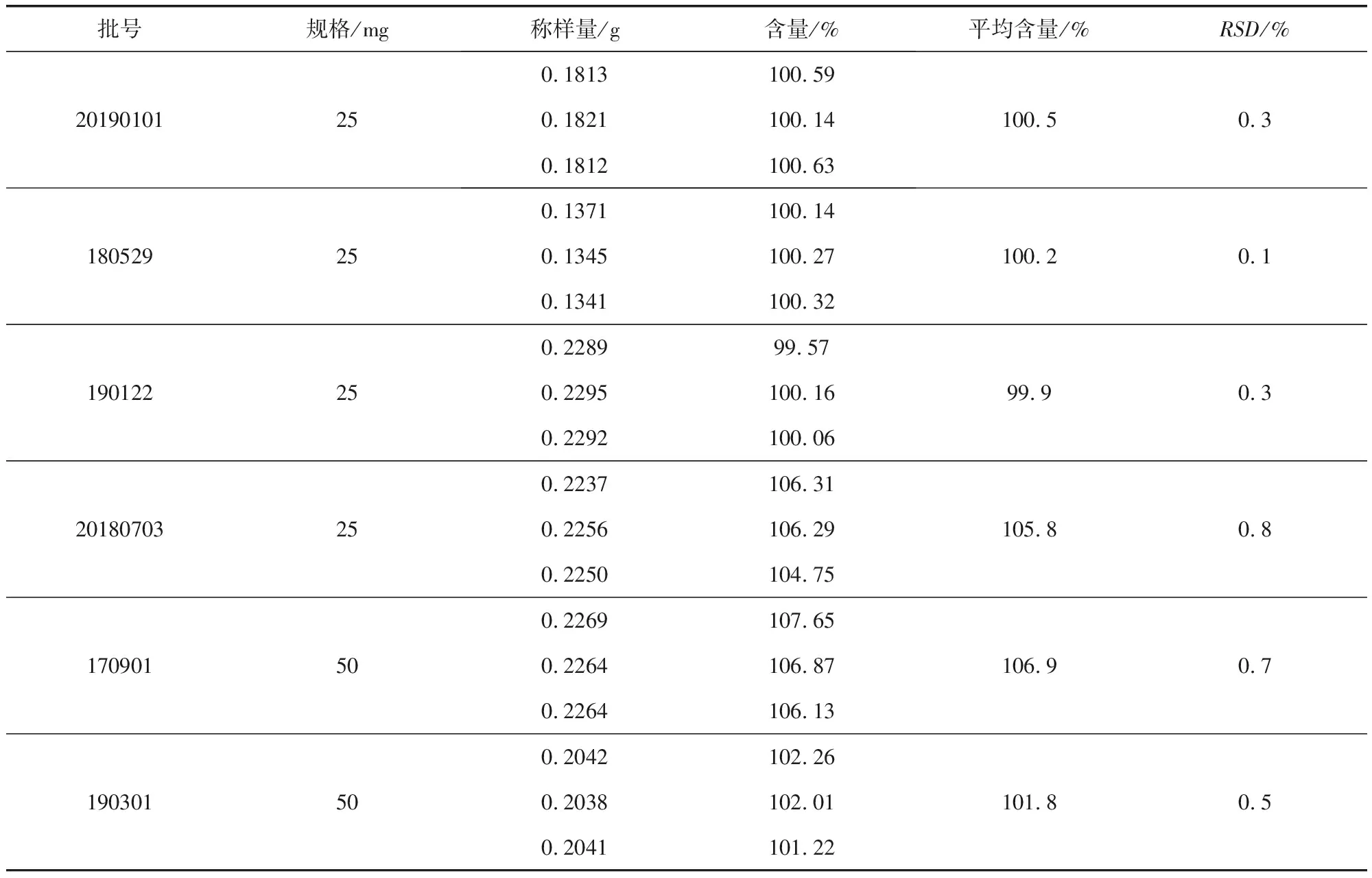
表3 液相测定结果Tab 3 Determination results of HPLC
3 讨论与结论
3.1 与容量分析结果比较 取6批次样品,按照《中国兽药典(2015年版)》一部中盐酸左旋咪唑片的含量测定项下的滴定法进行测定,结果见表4,并与高效液相色谱法比较,两者相对偏差均不超过1.0%,结果无显著差异。但滴定法存在以下问题:一是需使用毒性较大的三氯甲烷进行提取;二是步骤过长过多,实验误差较大;三是终点蓝色难以判断,容易过终点误读,导致实验结果偏高。相比较而言,高效液相色谱法更简便,也无需三氯甲烷、醋酐等危险化学试剂,更有利于实验的开展,实验结果也更为准确可靠。
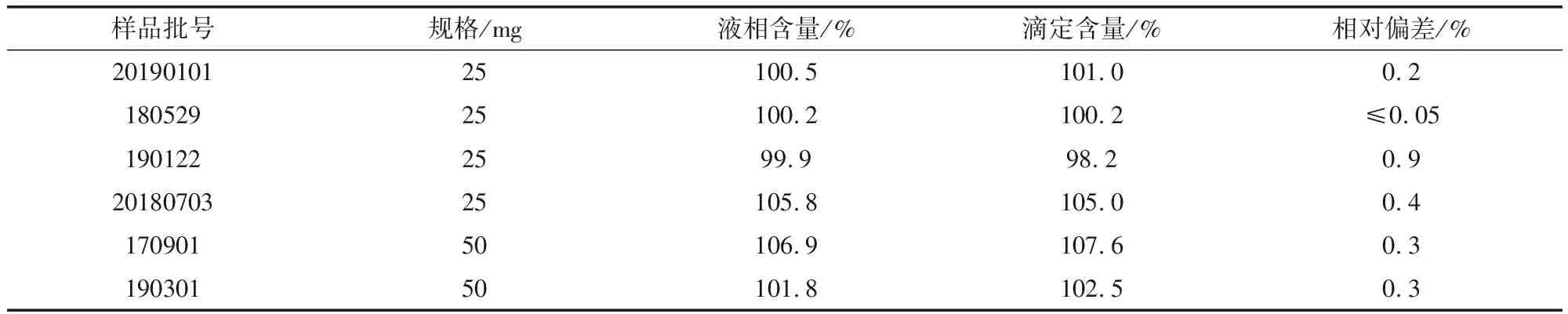
表4 液相含量测定与容量分析结果比较Tab 4 Comparisons between HPLC and volumetric analysis
3.1 提取方式的选择 在提取溶剂选择上,分别采用了65%甲醇、甲醇和流动相三种溶剂进行超声提取。结果表明纯甲醇提取后样品峰形较差,65%甲醇和流动相提取后的样品峰形较好,本文采用了流动相提取。在是否过滤的选择上,过滤与不过滤峰面积相对偏差为0.2%,对含量测定影响不大,由于过滤后溶液更澄清,更有利于第二步配制,本文采用了过滤的方法。在超声时间的选择上,实验过程中分别采用了超声5、10、20 min三种方式,峰面积分别为5255.33350,5262.27686,5247.13623,RSD为0.1%,因此选择超声时间为10 min。
3.2 检测波长的选择 对盐酸左旋咪唑溶液在190~400 nm波长范围内扫描,扫描结果表明在212 nm波长处有最大吸收,结合文献[5]-[6]报道中基本选择214 nm为检测波长,基于212和214 nm属于光谱扫描误差内,因此还是选择214 nm为检测波长,与文献保持一致。
3.3 流动相的选择 本文采用了甲醇-0.2%磷酸溶液以及乙腈-0.05 mol/L磷酸二氢钾溶液两种流动相配比进行试验,供试品溶液采用流动相进行稀释配制成50 μg/mL的溶液。结果表明,甲醇-0.2%磷酸溶液流动相下的盐酸左旋咪唑峰峰形不理想,峰形有较明显的拖尾现象,且有较明显的溶剂吸收峰,出峰时间在8 min左右,理论板数在8000左右。而乙腈-0.05 mol/L磷酸二氢钾溶液(三乙胺调节pH值至7.0)(20∶80,v/v)的流动相下的盐酸左旋咪唑峰峰形对称性良好,无干扰,无拖尾,出峰时间在13 min左右更为合理,无明显的溶剂吸收峰,理论板数在10000以上。因此选择后者作为流动相更适宜。
采用高效液相色谱法对盐酸左旋咪唑片中盐酸左旋咪唑的含量进行测定,对提取溶剂,提取方式和色谱条件进行了研究和优化,方法学验证结果表明该方法定量准确,与现行有效标准的含量结果一致,为盐酸左旋咪唑片的质量控制提供了更快捷、便利的操作方法。
