高效液相色谱法检测鸡可食性组织中的磺胺氯吡嗪残留
2019-09-06邢玉娟吕凤霞陈玲牛华星刘运镇邱树磊冯文丽刘联盟李灵娟
邢玉娟,吕凤霞,陈玲,牛华星,刘运镇,邱树磊,冯文丽,刘联盟,李灵娟
(1.江苏农牧科技职业学院,江苏泰州 225300;2.河南牧翔动物药业有限公司,郑州 451162;3.山东省兽药质量检验所,济南 250022)
磺胺氯吡嗪钠属于磺胺类广谱抗菌药,在兽医临床主要用于球虫病的治疗,此外还可用于球虫病暴发时继发禽伤寒、禽霍乱的防治[1-3],但过分滥用可导致食品中大量残留。目前,欧美国家及我国农业农村部规定了磺胺类药物在动物性食品中的最高残留限量为 0.1 mg/kg[4]。近年来对动物源性食品中磺胺类药物残留检测方法研究不断深入,主要包括免疫学方法、高效液相色谱法、气相色谱法、液质联用法、气质联用法等[5-8]。本试验对聂巧等[9]建立的鸡组织中磺胺氯吡嗪钠残留量的HPLC检测方法进行复核验证,以期对磺胺氯吡嗪钠的残留实施监控提供技术支持。
1 材料与方法
1.1 仪器与试剂
1.1.1 仪器 Waters 2695 高效液相色谱仪,2489uV/Vis二极管陈列检测器,美国Waters 公司;C18填料固相萃取小柱为美国Supelco 公司产品;PT1200组织匀浆机,瑞士KINEMATICA 公司;XH-J涡旋混合器,杭州浣熊仪器科技有限公司;N-EVAP34氮吹仪,Organomation Assciates公司;BS110S电子天平,德国赛多利斯天平公司;MXB22超声仪,华粤行仪器有限公司。
1.1.2 试剂与材料 乙腈、甲醇为色谱纯,天津市科密欧有限公司;其余试剂为分析纯。磺胺氯吡嗪钠对照品,含量99.0%,批号40908,购自德国Dr.Ehrenstorfer公司。
1日龄AA白羽肉鸡,30只,购自开封正大有限公司。使用不含任何药物的全价配合饲料饲养至30日龄,颈静脉放血处死,无菌采集胸肌、肝脏、肾脏和皮脂组织样品,组织匀浆后置于-20 ℃冰箱冰冻保存。
1.2 主要试液配制 0.02 mol/L磷酸二氢钾水溶液:准确称取2.7218 g磷酸二氢钾,用1000 mL超纯水定容并混匀,即制得0.02 mol/L磷酸二氢钾水溶液;5%氨化乙腈:准确吸取氨水5 mL,置于100 mL容量瓶中,用乙腈稀释至刻度,摇匀即得;乙腈-0.1 mol/L盐酸水溶液:将乙腈和0.1 mol/L盐酸水溶液按50∶50(V/V)比例混匀;乙腈饱和正己烷溶液:分别准确量取乙腈和正己烷各200 mL,充分混匀静置,上层液体即为乙腈饱和正己烷;流动相:将乙腈和0.02 mol/L磷酸二氢钾水溶液按35:65(V/V)比例混匀,经0.22 μm有机滤膜过滤,超声脱气20 min;磺胺氯吡嗪储备液:精密称取磺胺氯吡嗪钠对照品28.9 mg于25 mL容量瓶中,用甲醇溶解并定容,即为浓度为1000 μg/mL的磺胺氯吡嗪标准储备液。置-20 ℃冷冻保存;磺胺氯吡嗪标准工作液:准确吸取适量磺胺氯吡嗪标准储备液,用流动相稀释成适宜的磺胺氯吡嗪系列标准工作液。
1.3 色谱工作条件 流动相:乙腈-0.02 mol/L磷酸二氢钾水溶液(35:65,V/V);色谱柱:Waters Xbridge C18柱(4.6 mm×250 mm,5 μm);检测波长272 nm;流速1.0 mL/min;柱温35 ℃;进样量20 μL[9-11]。
1.4 标准曲线建立 准确量取适量磺胺氯吡嗪钠储备液,用流动相稀释,配制成含磺胺氯吡嗪钠质量浓度为0.08、0.20、1.00、2.00、10.00、20.00 μg/mL的标准溶液。供HPLC测定,记录图谱、采用外标法进行计算,以测得的峰面积A为纵坐标,相应质量浓度C为横坐标,绘制标准工作曲线,求出回归方程和相关系数(R2)。
1.5 样品前处理
1.5.1 样品的提取 称取2.00±0.01 g待测组织于50 mL离心管中,加入3 g无水硫酸钠,再加入5%氨化乙腈9 mL,旋涡震荡3 min,超声10 min后,离心15 min(4 ℃,12000 r/min),将上清液移至50 mL离心管中。残渣用上述相同方法重复提取一次,合并上清液。于上清液中加入5 mL乙腈饱和正己烷,旋涡振荡2 min,离心10 min(4 ℃,12000 r/min),弃去正己烷,保留下层液体备用,再重复该方法除脂2次。将除脂后液体于40 ℃水浴氮气吹至近干。用1 mL乙腈-0.1 mol/L盐酸水溶液(50∶50,V/V)的复溶液溶解近干残渣,旋涡振荡1 min;再加0.1 mol/L盐酸水溶液3 mL,旋涡振荡1 min,离心10 min(4 ℃,12000 r/min),取上清液备用,记为A液。残渣加0.1 mol/L盐酸水溶液3 mL涮洗离心管,旋涡振荡30 s,记为B液。
1.5.2 样品的净化 Oasis MCX(60 mg/3 mL)小柱依次用甲醇3 mL活化,超纯水3 mL平衡,将上清液A全部过柱;依次B溶液3 mL、甲醇1 mL淋洗;用5%氨化乙腈6 mL洗脱,收集洗脱液于10 mL离心管中,40 ℃水浴氮气吹干,用1 mL流动相溶解残渣,经0.22 μm有机滤膜过滤,供HPLC测定。
1.6 方法的灵敏度考察 取空白肝脏、肾脏、皮脂和肌肉组织样品添加适量浓度的磺胺氯吡嗪钠标
准工作液,各5个平行,按照样品前处理方法处理后检测,记录图谱。取信噪比(S/N)≥3时的样品浓度为检测限(LOD);以信噪比(S/N)≥10,且准确度与精密度符合要求的样品浓度为定量限(LOQ)。
1.7 方法的准确度和精密度考察 采用标准添加法,进行回收率试验。配制肌肉、肝脏、肾脏和皮脂在0.04、0.10、10.00 mg/kg三个添加浓度水平,按照样品前处理方法处理后检测。各浓度5个样品平行试验,重复3次,求添加回收率以及批内、批间的相对标准偏差。
2 结果与分析
2.1 色谱分离 在优化的色谱条件下,测得鸡肌肉、肝脏、肾脏、皮脂中磺胺氯吡嗪钠的保留时间分别为7.503、7.526、7.515、7.507 min,色谱峰峰形较佳,均为基线分离峰。鸡肌肉、肝脏、肾脏、皮脂空白提取液在上述时间均无干扰峰出现(图1)。
2.2 标准曲线 磺胺氯吡嗪钠标准溶液在0.08~20.0 μg/mL浓度范围内,药物浓度与峰面积呈良好的线性关系,标准工作曲线Y=109.56X-74.054,R2为0.9999。
2.3 灵敏度 鸡肌肉、肝脏、肾脏和皮脂组织的空白试料经前处理后检测,测定结果表明:在相应的保留时间,空白试料对所测分析物无干扰。添加浓度为40 μg/kg时信噪比(S/N)均大于10,信噪比满足定量限的要求,确定本方法的定量限。添加浓度为20 μg/kg时信噪比(S/N)均大于3,信噪比满足定量限的要求,确定为本方法的定量限。
2.4 准确度、精密度 样品添加回收率结果见表1。四种组织的回收率均在74.70%~93.67%,批内及批间变异系数均小于10%。表明该方法准确度、精密度高,重复性好。
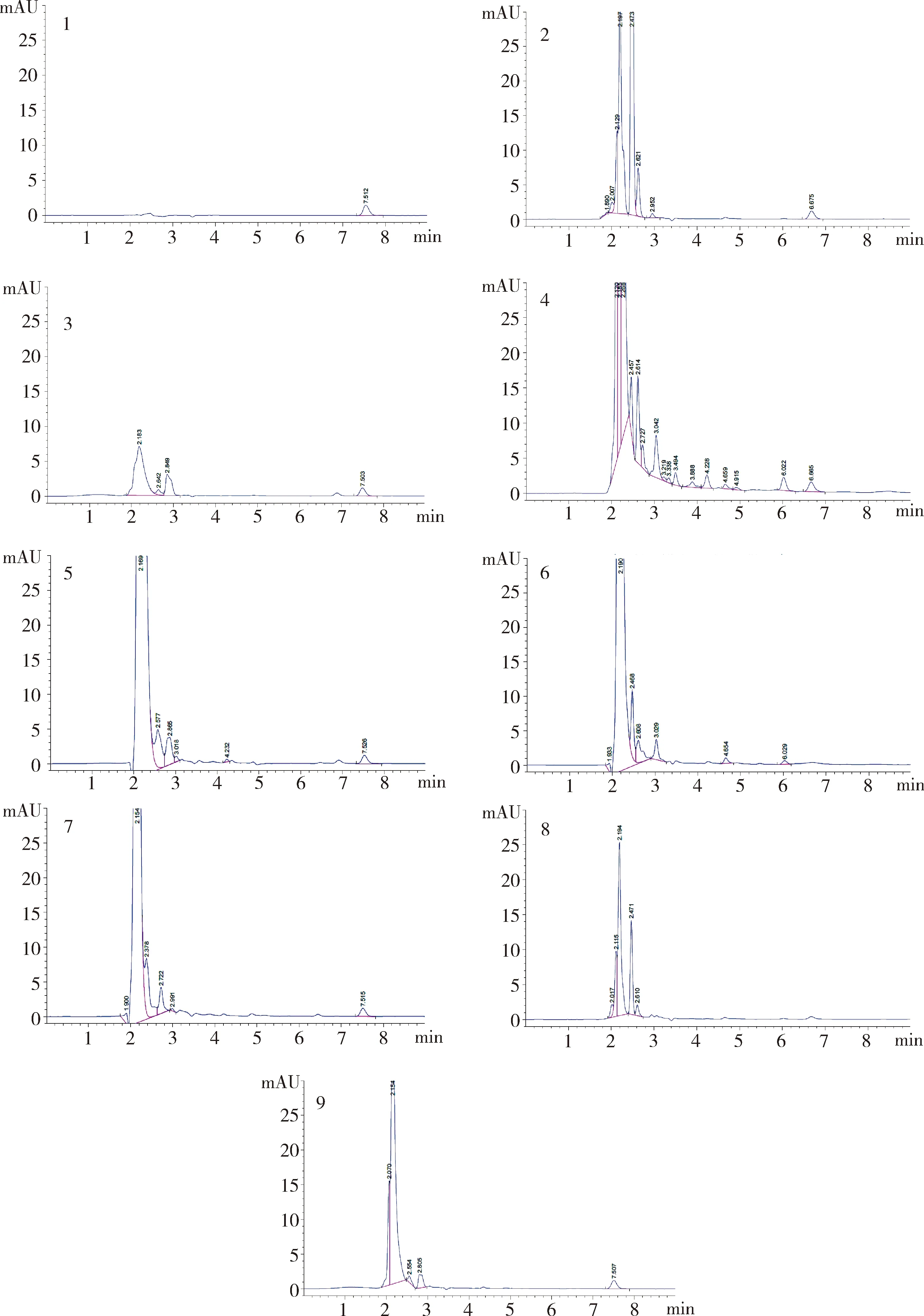
(1为0.2 μg/mL标准溶液;2为肌肉空白样品;3为100 μg/kg肌肉添加样品;4为肝脏空白样品;5为100 μg/kg肝脏添加样品;6为肾脏空白样品;7为100 μg/kg肾脏添加样品;8为皮脂空白样品;9为100 μg/kg皮脂添加样品)图1 鸡组织中添加磺胺氯吡嗪钠色谱图Fig 1 The chromatograms of sulfaclozine in chicken tissues
表1 回收率试验结果
Tab 1 Results of recovery tests
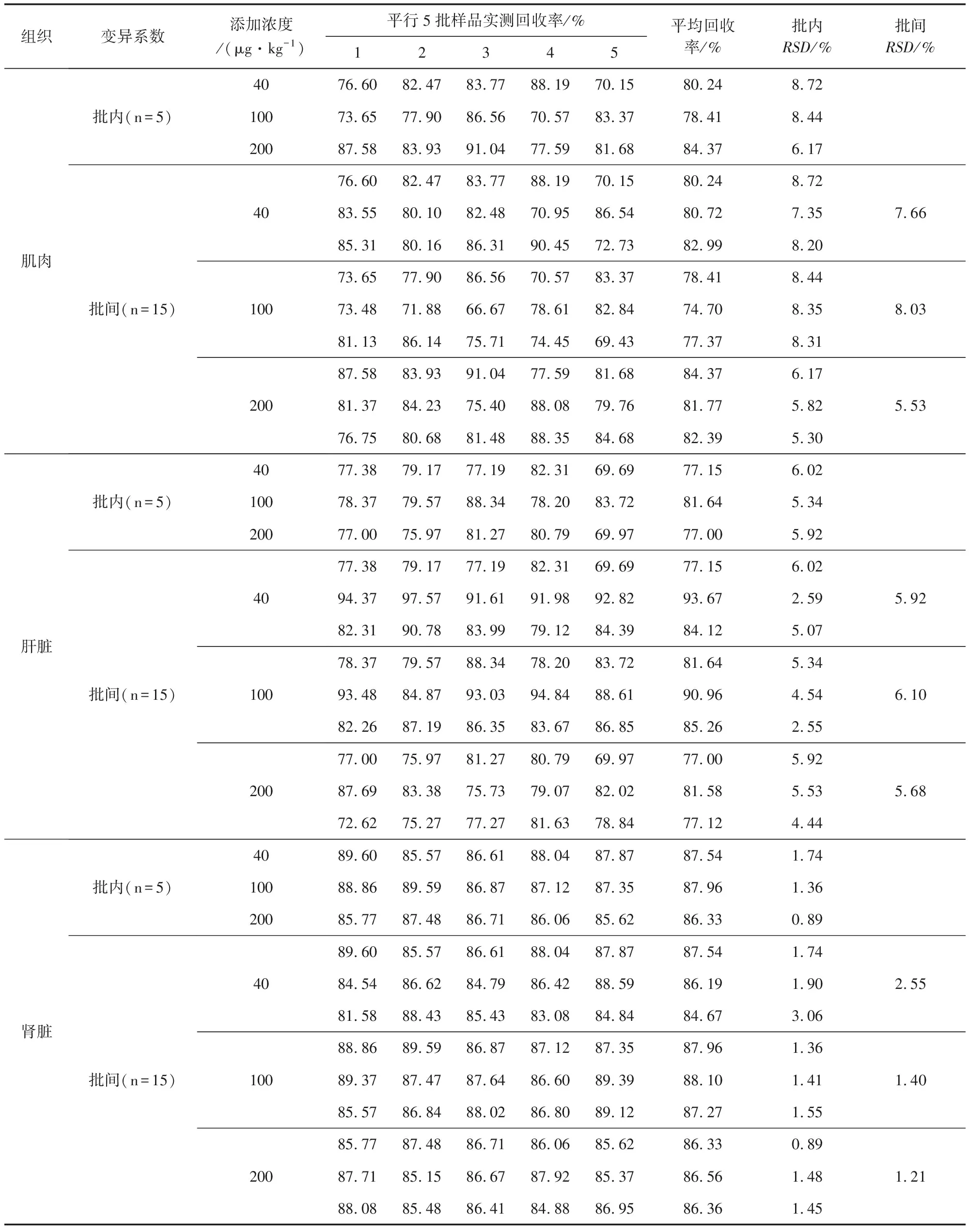
组织变异系数添加浓度/(μg·kg-1)平行5批样品实测回收率/%12345平均回收率/%批内RSD/%批间RSD/%肌肉批内(n=5)批间(n=15)401002004010020076.6082.4783.7788.1970.1580.248.7273.6577.9086.5670.5783.3778.418.4487.5883.9391.0477.5981.6884.376.1776.6082.4783.7788.1970.1580.248.7283.5580.1082.4870.9586.5480.727.3585.3180.1686.3190.4572.7382.998.2073.6577.9086.5670.5783.3778.418.4473.4871.8866.6778.6182.8474.708.3581.1386.1475.7174.4569.4377.378.3187.5883.9391.0477.5981.6884.376.1781.3784.2375.4088.0879.7681.775.8276.7580.6881.4888.3584.6882.395.307.668.035.53肝脏批内(n=5)批间(n=15)401002004010020077.3879.1777.1982.3169.6977.156.0278.3779.5788.3478.2083.7281.645.3477.0075.9781.2780.7969.9777.005.9277.3879.1777.1982.3169.6977.156.0294.3797.5791.6191.9892.8293.672.5982.3190.7883.9979.1284.3984.125.0778.3779.5788.3478.2083.7281.645.3493.4884.8793.0394.8488.6190.964.5482.2687.1986.3583.6786.8585.262.5577.0075.9781.2780.7969.9777.005.9287.6983.3875.7379.0782.0281.585.5372.6275.2777.2781.6378.8477.124.445.926.105.68肾脏批内(n=5)批间(n=15)401002004010020089.6085.5786.6188.0487.8787.541.7488.8689.5986.8787.1287.3587.961.3685.7787.4886.7186.0685.6286.330.8989.6085.5786.6188.0487.8787.541.7484.5486.6284.7986.4288.5986.191.9081.5888.4385.4383.0884.8484.673.0688.8689.5986.8787.1287.3587.961.3689.3787.4787.6486.6089.3988.101.4185.5786.8488.0286.8089.1287.271.5585.7787.4886.7186.0685.6286.330.8987.7185.1586.6787.9285.3786.561.4888.0885.4886.4184.8886.9586.361.452.551.401.21
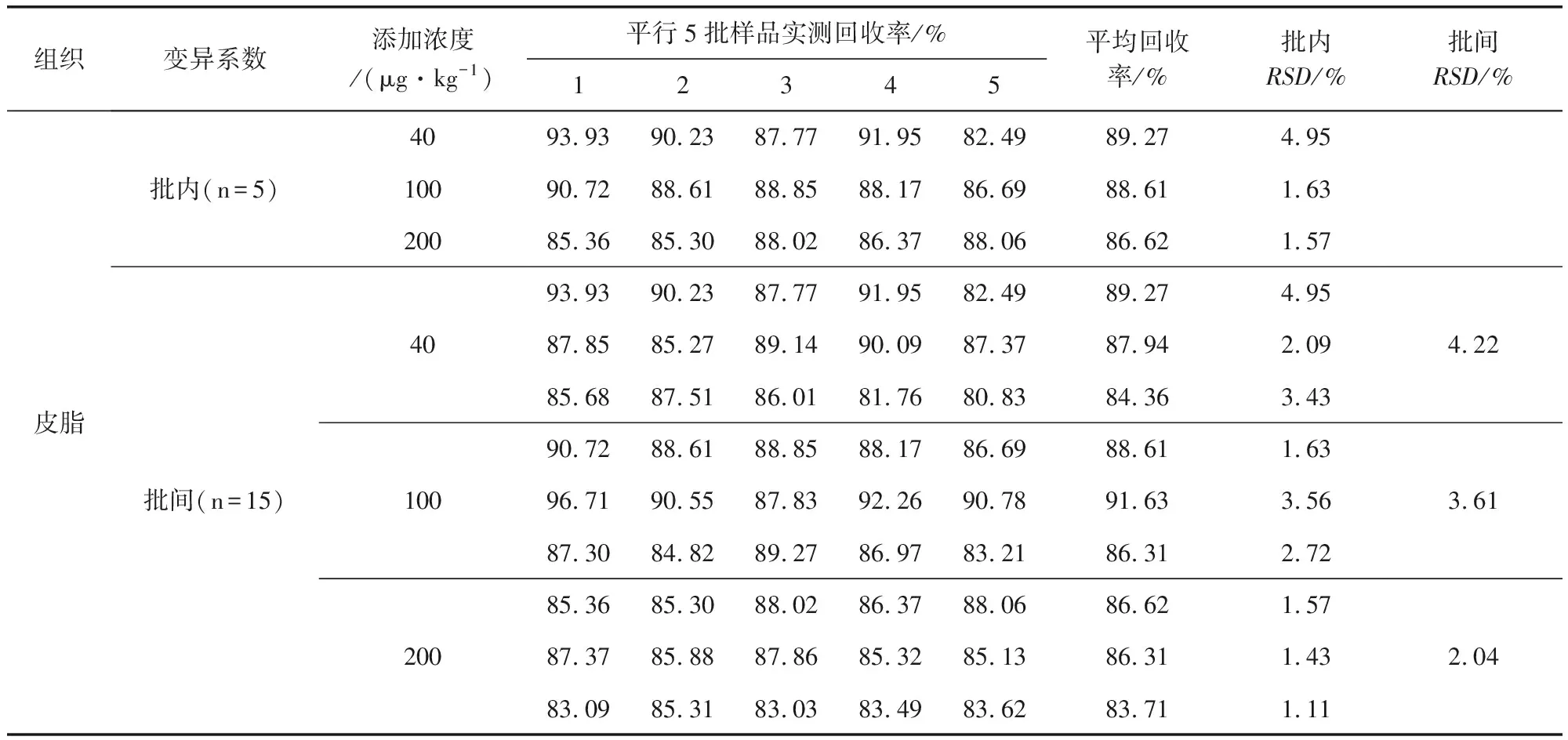
续表
3 讨论与结论
3.1 方法学的验证 本试验采用 HPLC 法测定鸡可食性组织中磺胺氯吡嗪钠的药物浓度。测定磺胺氯吡嗪钠在鸡可食性组织中的检测限为20 μg/kg,定量限为 40 μg/kg,与聂巧等建立的磺胺氯吡嗪残留钠的 HPLC 检测法研究结果一致[9]。在鸡肌肉、肝脏、肾脏、皮脂各组织中添加浓度分别为40、100、200 μg/kg时,各组织样品中磺胺氯吡嗪钠的平均回收率范围为74.70%~93.67%,高于聂巧等报道的75.46%~89.40%,批内和批间变异系数均小于9%,方法具有较好的回收率和精密度,证明该方法能对鸡可食性组织中磺胺氯吡嗪残留量进行有效的监控。
3.2 色谱条件的选择 本试验通过对磺胺氯吡嗪溶液进行紫外全波长扫描得知,在200~400 nm波长条件下,272 nm处有较大吸收,且在此条件下,组织样品无杂质干扰峰,故选择272 nm作为磺胺氯吡嗪的检测波长,柱温控制在35 ℃,磺胺氯吡嗪的检测取得较满意的效果。
试验过程中比较了甲醇-0.02 mol/L磷酸二氢钾水溶液、乙腈-0.017 mol/L磷酸水溶液、乙腈-0.02 mol/L磷酸二氢钾水溶液作为流动相,其中乙腈-0.02 mol/L磷酸二氢钾水溶液作为流动相时基线较其他两种流动相平稳。经过多次调整磷酸二氢钾的浓度和两者的比例,在比较了峰形、杂峰和药物峰分离等情况后,最终选择乙腈-0.02 mol/L磷酸二氢钾溶液(35∶65)作为检测的流动相[12],流速1.0 mL/min,磺胺氯吡嗪保留时间在7.5 min左右,得到的峰形良好,空白组织提取液在药物峰保留时间处无干扰峰出现。
3.3 提取方法的优化 MCX洗脱液有5%氨化乙腈、5%氨化甲醇等[13]。通过对比试验发现5%氨化甲醇洗脱后杂峰在药物峰附近有干扰,肝、肾组织表现尤为明显;而5%氨化乙腈净化效果及回收率理想。乙腈提取液可以避免从动物组织中提取过多的脂肪,而且乙腈具有良好的蛋白沉淀效果。用乙腈提取后,再用丙酮提取,磺胺氯吡嗪回收率在原有基础上提高了13%[14]。由于磺胺氯吡嗪不溶于正己烷,所以,在提取过程结束后再经正己烷进行脱脂处理,可以脱去脂肪,从而减少杂质的干扰[15]。样品提取过程中加入无水硫酸钠可以促进水相和有机相分离,同时硫酸钠的存在还可以促使蛋白质的变性分散,从而防止样品形成团块状而影响提取效率,以促进溶质的析出,提高了药物的提取率[16-17]。
在提取过程中若依靠旋转蒸发仪浓缩提取液,易出现蒸干后吸附于瓶壁的现象,降低样品回收率,故采用氮吹仪吹至近干[18],再用复溶液溶解,可减少损失。提取过程结束后,利用乙腈-0.1 mol/L盐酸溶液(50∶50)复溶液溶解残渣。复溶液的pH值可以改变药物的离子化程度,药物必须完全离子化才能保证药物完全保留于阳/阴离子交换柱上[19]。
本方法采用5%氨化乙腈提取鸡组织中的磺胺氯吡嗪,正己烷除脂,40 ℃水浴氮气吹至近干后采用复溶液(乙腈∶0.1 mol/L盐酸水溶液50∶50,V/V)和0.1 mol/L盐酸水溶液两次溶解,经MCX萃取柱净化,高效液相色谱仪检测,验证了鸡组织中磺胺氯吡嗪残留量的检测方法。磺胺氯吡嗪在鸡肌肉、肝脏、肾脏和皮脂中的定量限为40 μg/kg,鸡空白组织磺胺氯吡嗪添加样品在40~200 μg/kg浓度范围内,回收率在74.70%~93.67%之间。整个实验过程简单、快速、回收率较高,适合鸡组织中磺胺氯吡嗪残留的定量检测。
