手性紫罗兰酮生物碱衍生物Ion-31a的制备方法优化
2019-09-03吴潇然聂江平段宏泉
樊 晔 ,吴潇然 ,聂江平 ,秦 楠 ,2,段宏泉 ,2
(1.天津医科大学药学院生药学教研室,天津市临床药物关键技术重点实验室,天津300070;2.天津医科大学基础医学研究中心,天津300070)
恶性肿瘤是我国以及全球主要的公共健康问题,在我国肿瘤死亡约占全部死因的1/4[1],根据国际癌症研究中心发布的数据显示,2012年全球恶性肿瘤新发病例约1 409万,死亡约820万[2],在恶性肿瘤中,乳腺癌是导致死亡的第二大肿瘤。在大量临床病例中,肿瘤转移是导致恶性肿瘤患者死亡的最主要原因,约90%恶性肿瘤患者的死亡原因与肿瘤转移相关[3]。在转移早期阶段,乳腺癌细胞分泌蛋白水解酶,从而使它们从原发位点分离并退化细胞外基质,从而刺激癌细胞与多种方向反应而转移,以促进肿瘤细胞的进程,在血液中循环后,癌细胞外渗入他们能存活和增殖的远端器官而实现转移[4-5]。本课题组前期在湖北省鄂西地区土家族常用民间药材中发现转筋草具有抗肿瘤转移活性。转筋草乙醇提取物分离提纯后分离得到三萜、甾体、生物碱等各类成分,并对各类化合物进行抗肿瘤转移的活性筛选,发现其中生物碱化合物具有抗肿瘤转移活性[6],其中一类结构新颖的紫罗兰酮生物碱对乳腺癌细胞MDA-MB-231趋化、迁移、侵袭能力具有显著抑制作用,对人脐静脉内皮细胞(HUVEC)的体外迁移及小管形成有明显抑制作用[7]。以该化合物为先导化合物,经过结构优化得到了活性显著提高的紫罗兰酮生物碱衍生物Ion-31a(图1),并发现此衍生物通过下调肿瘤细胞转移相关的重要信号分子整合素-β(intergrinβ)、蛋白激酶 C-ξ(PKCξ)的磷酸化水平而发挥抗肿瘤转移作用[8]。化合物Ion-31a活性突出,具有非常好的新药开发价值,但原文献报道的合成路线复杂,产率很低,本文通过建立新的合成路线,优化反应条件以得到提高Ion-31a的总收率,同时保证其良好的光学纯度。
1 材料与方法
1.1 仪器与试剂 SHB-ⅢA循环水式多用真空泵;W201恒温浴锅;低温冷却液循环泵(上海豫康科仪器设备有限公司);DF-101S集热式恒温加热磁力搅拌器;Advance III 400M核磁共振仪(Bruker公司,TMS为内标);DZX-3型真空干燥箱;ZF-I型三用紫外分析仪;EYELA PSL-1810型低温反应器;KQ-500B超声波清洗器;青岛海洋高效薄层层析硅胶板 GF254(200 mm×200 mm);202电热恒温干燥箱;N-1100旋转蒸发仪。α-紫罗兰酮纯度为99%,购买于希尔贝思(天津)科技有限公司;柱色谱和薄层色谱用硅胶均系青岛海洋化工厂生产,所用试剂均为分析纯;氘代试剂ALDRICH公司生产。
1.2 优化后的Ion-31a的合成工艺 见图1。
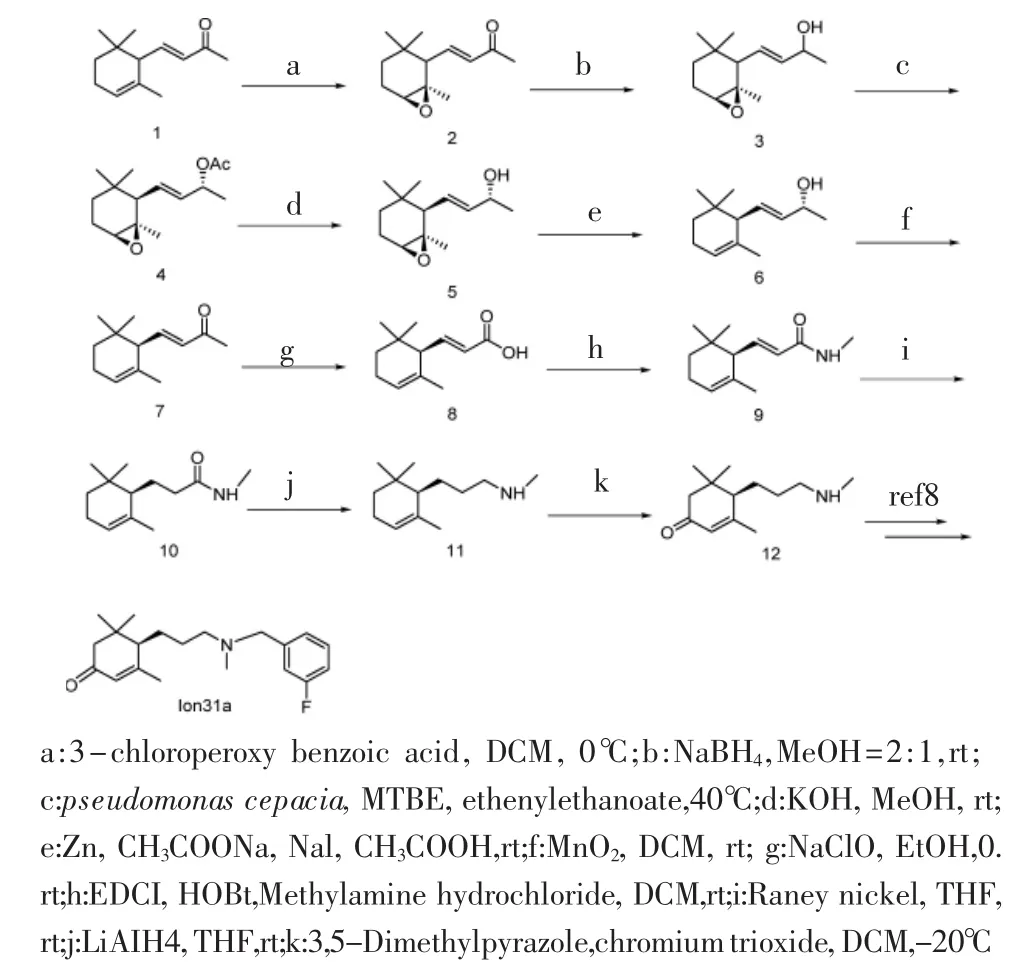
图1 化合物Ion-31a的合成路线Fig 1 Synthetic route of compound Ion-31a
1.3 化合物合成方法
1.3.1 化合物4的合成 取1当量化合物3于反应瓶中,加入适量甲基叔丁基醚溶解,再加入1当量洋葱假单胞菌脂肪酶(pseudomonas cepacia)和12当量醋酸乙烯酯。之后将反应瓶于40℃油浴条件下搅拌 1 周,TLC 监测反应完全(PE:EA=4∶1,V/V)。将反应液离心,取上清液旋干得粗品,然后将粗产品经硅胶柱色谱(PE∶EA=10∶1,V/V)得化合物 4。
1.3.2 化合物6的合成 取1当量化合物5粗品置于反应瓶中,用适量乙酸溶解,在室温下加入1.1当量锌粉和3当量乙酸钠,搅拌5 min后加入2当量碘化钠,室温反应过夜,TLC监测反应完全(PE:EA=10:1,V/V)。将反应液用少量EA稀释,用饱和碳酸氢钠溶液将pH调至中性,再用EA萃取,收集有机相加入无水硫酸镁干燥,旋干,得到化合物粗品,然后将粗产品经硅胶柱色谱(PE∶EA=25∶1,V/V),得化合物6。
1.3.3 化合物10的合成 取1当量化合物9于反应瓶中,用THF溶解,加雷尼镍混合物(50%在水中)氢气置换后,向反应瓶中持续通入氢气在室温下搅拌 1 h,TLC 监测反应完全(PE∶EA=3∶2,V/V)。反应完成后,将反应液用硅藻土抽滤,旋干,即得化合物10。
1.3.4 化合物11的合成 称取4当量四氢铝锂于反应瓶中,加入无水四氢呋喃,氮气保护于冰浴下搅拌,缓慢滴加1当量化合物10的无水四氢呋喃溶液,将反应液升温至30℃搅拌24 h。TLC监测反应完全(PE:EA=1∶1,V/V)。冰浴下缓慢滴加加入蒸馏水,4 mol/L氢氧化钠水溶液,反应液变成乳白色混悬液后,抽滤,浓缩滤液得粗品11。
1.3.5Ion-31a的合成 称取1当量化合物12于反应瓶中,用适量二氯甲烷使其溶解,加入1.5当量无水碳酸钾和1当量3-氟溴苄,在室温下搅拌20min后,TLC 监测反应完全(PE:EA:二乙胺=5:1:1,V/V)。浓缩溶剂得粗品,粗品经硅胶柱色谱(PE:EA=3:1,V/V)得Ion-31a。
2 结果
2.1 关键化合物的结构表征
2.1.1 化合物7的结构表征 黄色油状物,产率为28.8%。1H NMR(400MHz,CDCl3),δ6.61(dd,J=15.2 Hz,10.0 Hz,1H),6.03 (dd,J=15.2 Hz,6.1 Hz,1H),5.50(m,1H),2.34(s,3H),2.02(m,2H),1.61(m,3H),1.45(m,1H),1.13(m,1H),0.80(s,3H),0.72(s,3H).13CNMR(100MHz,CDCl3):δ149.1,132.7,59.6,59.1,52.1,30.8,28.9,28.1,27.0,23.9,21.9,21.4,20.4.
2.1.2 化合物8的结构表征 黄色油状物,产率为80%。1HNMR(400MHz,CDCl3),δ6.92(dd,J=15.2Hz,9.8Hz,1H),5.81(d,J=15.2Hz,1H),5.50(br s,1H),2.31(d,J=9.8Hz,1H),2.04(br s,2H),1.57(s,3H),1.47(m,1H),1.21(m,1H),0.93(s,3H),0.86(s,3H).13C-NMR(100MHz,CDCl3):δ171.4,152.9,131.6,122.8,121.5,54.1,32.5,31.1,27.7,26.8,23.0,22.8.
2.1.3 化合物9的结构表征 黄色油状物,产率为72%。1HNMR(400MHz,CDCl3),δ6.64(dd,J=15.2Hz,9.7Hz,1H),6.61(br s,1H)5.83(d,J=15.2Hz,1H),5.45(br s,1H),2.85(d,J=4.8Hz,3H),2.23(d,J=9.7Hz,1H),2.01(br s,2H),1.55(s,3H),1.46(m,1H),1.18(m,1H),0.90(s,3H),0.84(s,3H).13C-NMR(100MHz,CDCl3):δ166.8,144.6,132.3,124.8,122.1,53.8,32.3,31.2,27.6,26.8,26.2,23.0,22.8.
2.1.4 化合物10的结构表征 黄色油状物,产率为 95%。1H NMR(400MHz,CDCl3):δ5.47(br s,1H),5.33(m,1H),2.80(s,3H),2.21(t,2H),2.0-1.85(m,3H),1.68(s,3H),1.48(m,2H),1.45-1.10(m,2H),0.93(s,3H),0.87(s,3H).13C-NMR(100MHz,CDCl3):δ173.8,135.6,120.9,48.7,36.7,32.6,31.5,27.6,27.5,26.6,26.2,23.5,23.0.与文献报道[9]一致。
2.1.5Ion-31a的结构表征 黄色油状物,产率为52.1%。1H NMR(400MHz,CDCl3):δ7.25(m,1H,Ar-H),7.04(m,2H,Ar-H),6.92(m,1H,Ar-H),5.81(br s,1H),3.45(br s,2H),2.36(d,J=17.2Hz,1H),2.33(m,2H),2.17(s,3H),2.02(d,J=17.2Hz,1H),1.97(s,3H),1.84(m,1H),1.71(m,1H),1.57(m,2H),1.41(m,1H),1.04(s,3H),1.00(s,3H).13C NMR(100MHz,CDCl3):δ199.4,165.6,163.0(d,J=243.7Hz),142.0(d,J=6.9Hz),129.6(d,J=8.1Hz),125.1,124.3(d,J=2.7Hz),115.6(d,J=21.1Hz),113.8(d,J=21.1Hz),62.0,57.7,51.2,47.2,42.3,36.3,28.8,27.9,27.3,27.2,24.7.
2.2 目标化合物制备方法优化 关键中间体8的制备方法优化结果见表1。目标产物Ion-31a的制备方法优化结果见表2。

表1 不同拆分方法可得到中间体8的收率Yield of intermediate 8 obtained by different resolution methods
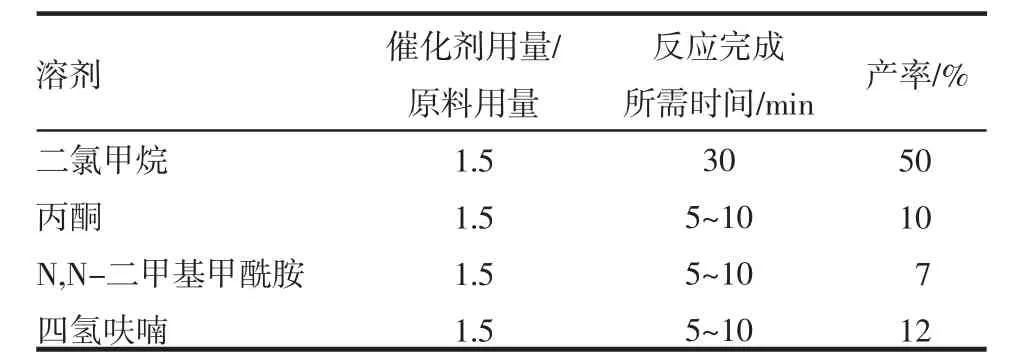
表2 不同溶剂对最后一步制备Ion-31a的产率影响Tab 2 Effects of different solvents on the yield of Ion-31a prepared in the last step
3 讨论
中间体8是整个路线合成的重要光学中间体,而对其合成方法的优化是Ion-31a合成路线优化的主要工作。化合物8合成的关键步骤是对外消旋原料进行拆分以得到单一手性的中间体。原路线是由消旋α-紫罗兰酮合成化合物8的外消旋体,再与(R)-(+)-1-苯乙胺成盐后使用结晶法对其进行拆分,得到R构型的中间体8。这也是分离手性化合物最经典、最常用的方法,广泛应用于大多数手性药物制备。其原理是外消旋体的化学性质使其与某一光学拆分剂反应以生成两种非对映体的盐,然后利用两者之间的溶解度和结晶速率的差异,通过结晶法进行分离,最后再脱去拆分剂,即得到单一构型的异构体。由于本文所需关键中间体8为油状物,结晶性较差,这可能在很大程度上限制了应用结晶法对其进行拆分,实际操作中发现,需对其进行成盐结晶操作,且得到的产物光学纯度不高,拆分损失较大,是影响目标化合物Ion-31a总产率的最主要原因。
根据文献报道,通过酶促拆分法,利用洋葱假单胞菌脂肪酶对仲羟基的选择性乙酰化,可得到光学纯的R构型α-紫罗兰酮类化合物[10]。如表1所示,由于酶促反应具有高度的立体选择性,使用酶催化的该步反应得到的产物光学纯度较高且可大量制备。使用酶催化反应合成单一构型化合物,再经简单的水解、氧化、卤仿反应得到高光学纯度的R构型化合物8。此步骤之后所有反应均不涉及化合物6位手性中心,因此该手性碳不会发生构型改变,不影响目标产物Ion-31a的光学纯度。因此,酶促拆分法较结晶拆分法获得中间体8优势明显,产率显著提高。
原文献报道的化合物11合成方法是使用四氢铝锂直接对化合物9的共轭不饱和酰胺进行还原得到化合物11。在进行还原反应时除生成目标化合物11以后,还会得到双键未被还原的副产物,这使得此反应产率较低,产物只有40%。
针对该反应的副产物情况,我们尝试分步还原,即先用雷尼镍催化氢化反应还原共轭不饱和酰胺的双键得到中间体10[11-12],再进行酰胺还原得到中间体11,该方法与原方法相比产率直接提高了30%。
