高压超声波协同改性红松松仁膜衣膳食纤维工艺优化及结构分析
2019-09-02王秋阳徐红艳
王秋阳, 付 刚, 王 超, 张 敏, 徐红艳
(延边大学,吉林 延吉 133002)
红松(Pinuskoraiensis)在中国东北的小兴安岭到长白山一带分布较广[1]。红松仁富含单不饱和脂肪酸和生育酚、蛋白质[2-3],具有良好的风味、营养和保健特性,深受消费者喜爱。红松松仁膜衣为松仁外层所附的一层薄皮,在坚果企业精加工松仁过程中,产生大量膜衣副产物[4]。课题组前期研究发现,红松松仁膜衣含有黄酮类化合物、多酚和蛋白质等成分[5],具有较高的营养价值。
膳食纤维(Dietary Fiber,DF)的溶解度与其化学结构关系密切,可分为水溶性膳食纤维(Soluble Dietary Fiber,SDF)和水不溶性膳食纤维(Insoluble Dietary Fiber,IDF)2种[6]。高膳食纤维饮食,可降低各种慢性疾病的风险[7-8],为人类健康的可持续性提供重要的健康益处,增加最终产品的营养价值。因此,有关膳食纤维的粒度、表面积、微观结构、改善其功能特性,以及提高在食品工业中的应用受到广泛关注[9]。本文采用高压-超声波协同法通过高温、高压以及高频振动的剪切作用使其中的非共价键发生变化从而改变其结构,达到改性目的。
本文以PIDF为原料,通过响应面法优化高压-超声波协同作用的改性条件,并采用扫描电子显微镜、X射线衍射、傅里叶变换红外光谱进行进一步分析。旨在为松仁膜衣的精深加工提供理论依据,促进其在食品、保健品领域的应用,为坚果副产物的综合利用提供新思路。
1 材料与方法
1.1 材料与试剂
红松松仁膜衣:梅河口市实全土特产品有限公司于2018年提供。
高峰α-淀粉酶(≥4 000 U/g),购自上海源叶生物科技有限公司;无水柠檬酸,购自潍坊英轩实业有限公司;柠檬酸三钠,购自福晨(天津)化学试剂有限公司;H2O2、无水乙醇等试剂均为国家级分析纯。
1.2 仪器与设备
PHS-3C型酸度计(上海仪电科学仪器股份有限公司);KQ-500DE型超声波清洗器(昆山市超声仪器有限公司);YXQ-LS-50SⅡ型立式压力蒸汽灭菌器(上海博讯实业有限公司医疗设备厂);D/max-2200型X射线衍射仪(日本理学公司);SU8010型扫描电子显微镜(日立仪器有限公司)。
1.3 PIDF的制备
根据GB 5009.88-2014方法,将红松松仁膜衣经粉碎后过60目筛,按料液比 1∶10(g/mL)加入蒸馏水,调节溶液pH值至中性,加入木瓜蛋白酶0.6 g,50 ℃水浴60 min;再调节溶液pH值至4.5,加入1%葡萄糖苷酶,60 ℃水浴30 min,灭酶10 min后抽滤,将滤渣烘干称重并粉碎,过60目筛得PIDF。
1.4 高压-超声波协同法改性PIDF的工艺流程
PIDF→按不同的料液比加入不同pH值1% H2O2溶液→以固定条件,即压力0.1 MPa、温度121 ℃、时间20 min进行处理→不同功率、时间的超声波处理→溶液调至中性→抽滤→乙醇醇沉→沉淀干燥→改性SDF[10]
1.5 响应面优化高压-超声波协同法改性PIDF工艺条件
考虑到节省原料及试剂等因素,因此选择pH值、超声波功率和超声波时间3个因素,采用Box-Behnken中心设计原理[11-13],设计高压-超声波协同法改性的响应面试验。试验因素和水平编码见表1[14-15]。
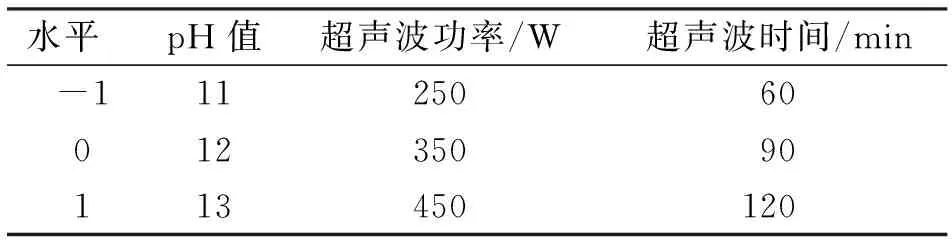
表1 高压-超声波协同法响应面试验因素水平
1.6 扫描电子显微镜、X射线衍射和傅里叶变换红外光谱测试
根据曹龙奎等[16]的方法对未改性SDF和改性SDF进行超微结构、X射线衍射强度以及红外光谱透光率的测定。
2 结果与分析
2.1 高压-超声波协同法改性PIDF单因素
2.1.1 pH值对改性SDF得率的影响
称取5.00 g PIDF,按1∶10(g/mL)加入不同pH值的1%过氧化氢溶液,在压力0.1 MPa、温度121 ℃条件下处理20 min,然后在超声波功率300 W,超声温度60 ℃条件下处理90 min,调节pH值至中性,醇沉后得到的沉淀烘干称重,计算改性SDF得率。
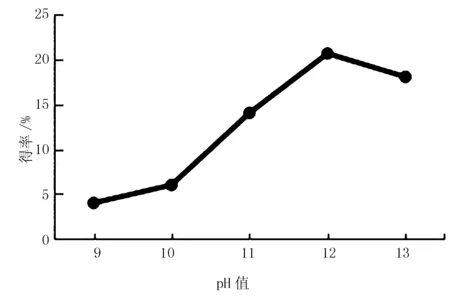
图1 pH值对红松松仁膜衣改性SDF得率的影响
pH值是影响膳食纤维溶解度和稳定性的重要因素。由图1可知,改性SDF得率随pH值的升高而逐渐增加,可能是体系中离子浓度升高,有利于膳食纤维充分溶解。当pH值为12时,改性SDF得率达到最高,达到饱和状态,为20.6%。当pH值超过12后,改性SDF结构被破坏并降低其溶解度,改性SDF得率下降[10]。因此,确定12为最适pH值。
2.1.2 超声波功率对改性SDF得率的影响
称取5.00 g PIDF,按1∶10(g/mL)加入pH值为12的1%过氧化氢溶液,在压力0.1 MPa、温度121 ℃条件下处理20 min,然后在不同超声波功率下,超声温度60 ℃处理90 min,调节pH值至中性,醇沉后得到的沉淀烘干称重,计算改性SDF得率。
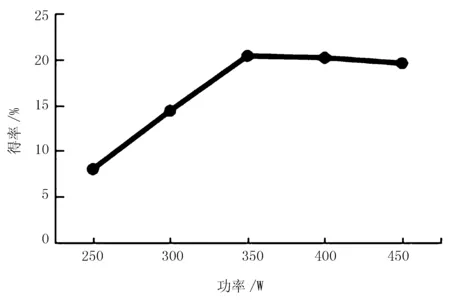
图2 超声波功率对红松松仁膜衣改性SDF得率的影响
由图2可知,当超声波功率由250 W上升到350 W时,改性SDF得率由6.2%增加到20.4%。当超声波功率为350 W时,其高频振动作用趋于饱和,改性SDF得率达到最大。当超声波功率超过350 W后,改性SDF得率略微降低。表明在相同时间内,超声波产生的空化和机械振动作用[17],使改性SDF中化学键断裂,导致得率下降,因此,确定350 W为最适超声波功率。
2.1.3 超声波时间对改性SDF得率的影响
称取5.00 g PIDF,按1∶10(g/mL)加入pH值为12的1%过氧化氢溶液,在压力0.1 MPa、温度121 ℃条件下处理20 min,然后在超声波功率350 W,超声温度60 ℃条件下分别处理不同时间,调节pH值至中性,醇沉后得到的沉淀烘干称重,计算改性SDF得率。
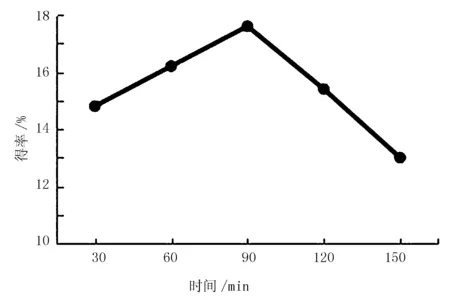
图3 超声波时间对红松松仁膜衣改性SDF得率的影响
由图3可知,当超声波时间从30到90 min时,改性SDF得率上升较快,当超声波时间为90 min时,改性SDF得率为17.6%,达到最大。时间超过90 min时,反应体系温度逐渐升高,改性SDF中的成分被分解成小分子,得率降低。因此,确定90 min为最适超声波时间。
2.1.4 料液比对改性SDF得率的影响
称取5.00 g PIDF,分别按1∶15、1∶20、1∶25、1∶30、1∶35(g/mL)加入pH值为12的1%过氧化氢溶液,在压力0.1 MPa、温度121 ℃条件下处理20 min,然后在超声波功率350 W,超声温度为60 ℃条件下处理90 min,调节pH值至中性,醇沉后得到的沉淀烘干称重,计算改性SDF得率。
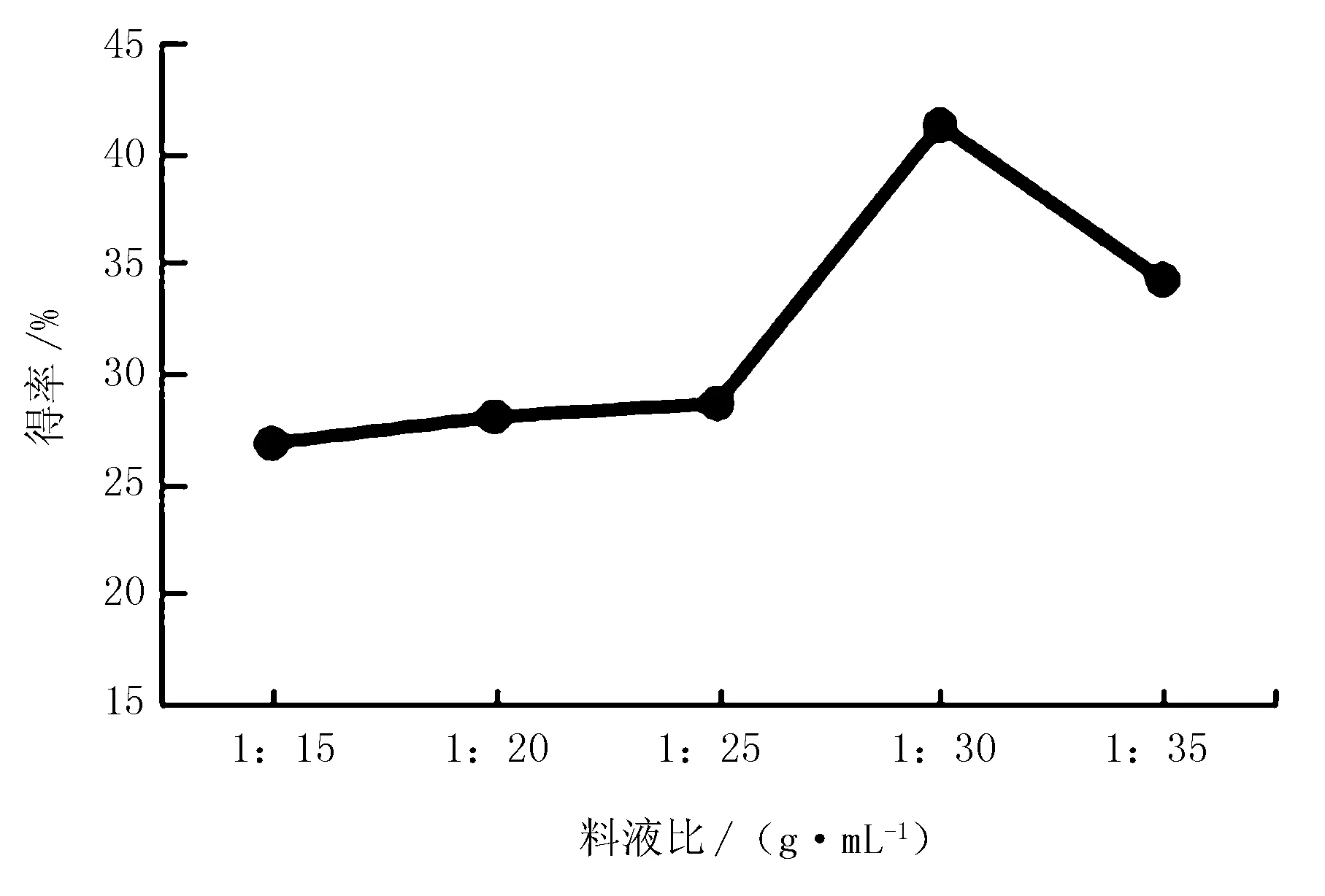
图4 料液比对红松松仁膜衣改性SDF得率的影响
由图4可知,当料液比从1∶15到1∶30时,改性SDF得率也随之增加,可能因为料液比过小时,原料不能在缓冲溶液中充分溶胀,影响了超声波与其相互作用,不能达到理想的改性效果;当料液比为1∶30时,改性SDF得率达到最大,为41.2%。之后料液比增加导致原料被稀释,改性效率降低,得率减少。因此,确定1∶30为最适料液比。
2.2 高压-超声波协同法改性PIDF响应面
2.2.1 响应面设计与结果
选取pH值、超声波功率和超声波时间3个试验因素进行响应面分析,以确定高压-超声波协同改性的最优条件。试验设计与结果见表2,系数检验见表3,方差分析见表4。
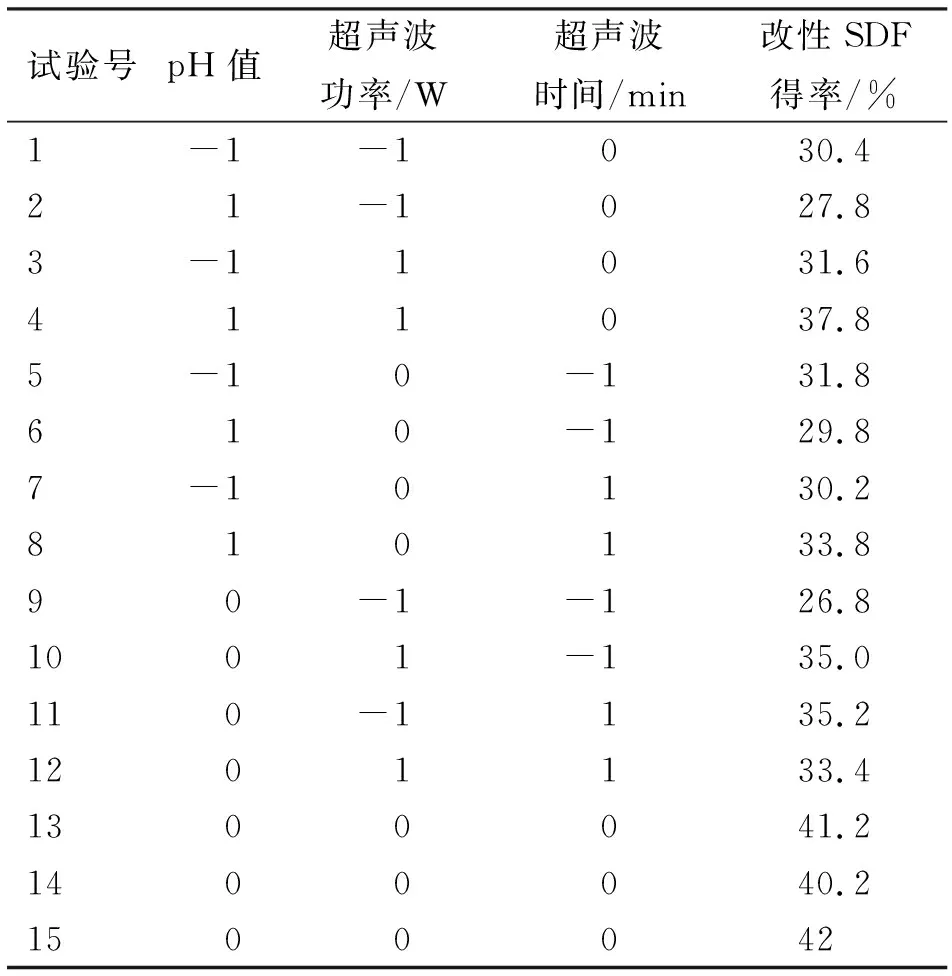
表2 响应面试验设计方案与结果
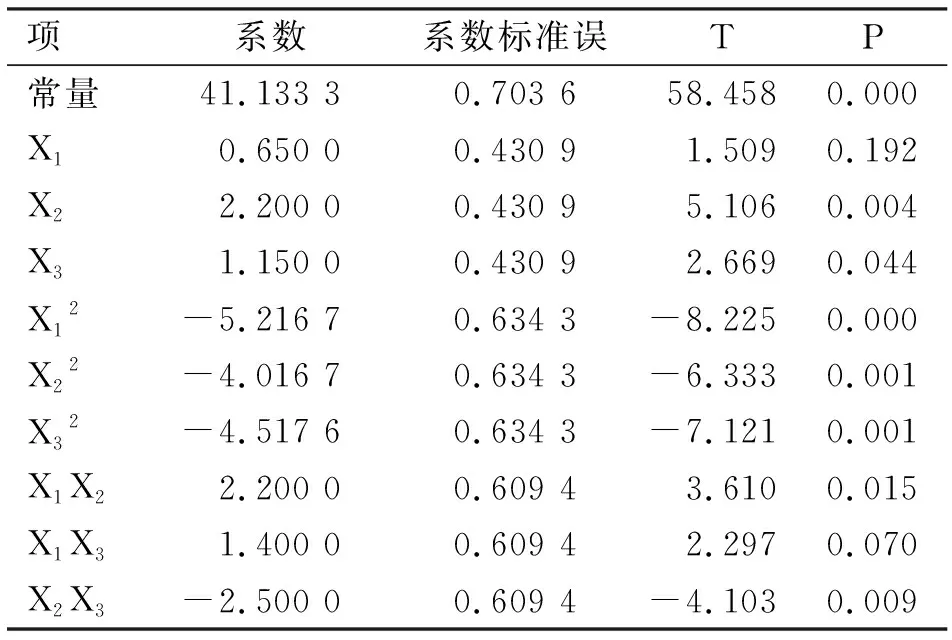
表3 响应面分析优化后改性SDF得率模型的系数检验
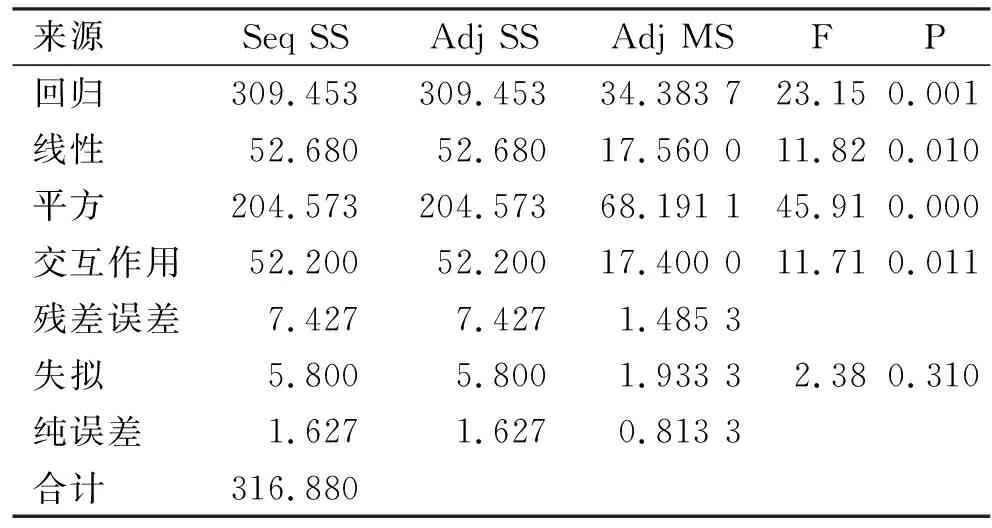
表4 响应面优化改性SDF得率回归方程的方差分析
改性SDF得率的结果经回归拟合,得到回归模型为:
Y=41.133 3+0.650 000X1+2.200 00X2+1.150 00X3-5.216 67X12-4.016 67X22-4.516 67X32+2.200 00X1X2+1.400 00X1X3-2.500 00X2X3
综上所述,对于年龄≥80岁患者,在3 h溶栓时间窗内,低剂量与标准剂量阿替普酶静脉溶栓具有相似的有效性和安全性。但是,本研究纳入样本较少,结论可能存在偏倚。因此,有待扩大样本量,反复论证低剂量阿替普酶治疗高龄急性脑梗死患者的疗效及安全性。
通过方差及其相关系数的可靠性分析模型,由表3可知,一次项X2、二次项X12、X22、X32和交互项X2X3极显著影响可溶性膳食纤维得率(P<0.01),一次项X3和交互项X1X2显著影响膳食纤维得率(P<0.05),其余项影响不显著(P>0.05)。
由响应面分析结果可知,R2=93.44%;膳食纤维得率回归模型的P值极显著(P<0.01);失拟项的P值不显著(P>0.05);表明该模型对各因素与响应值之间的真实关系具有可靠性,可以利用此模型进行分析和预测高压-超声波协同法对PIDF的改性效果。
2.2.2 高压-超声波协同法改性最优条件的确定
pH值、超声波功率和超声波时间3个因素影响改性SDF得率的响应面见图5。

图5 各因素交互作用的响应面图
综合分析响应面改性SDF得率的试验结果表明,各因素对改性SDF得率的影响大小排序为:超声波功率>超声波时间>pH值,其中,超声波功率影响极显著(P<0.01),超声波时间影响显著(P<0.05),交互项按影响大小排列依次为:超声波功率和超声波时间>超声波功率和pH值>pH值和超声波时间。
依据回归方程,最佳改性工艺为:pH值为12、超声波功率378 W、超声波时间92 min。
2.2.3 验证试验
根据最佳工艺条件进行3次验证试验,改性SDF得率分别为43.2%、42.6%和44.2%,平均得率为43.3%,验证试验结果与理论预测值41.53% 接近,RSD为1.8%,表明该模型符合实际情况,响应面法优化得到的高压-超声波协同改性条件具有真实可靠性。
2.3 高压-超声波协同法改性前后SDF结构分析
2.3.1 高压-超声波协同法改性前后SDF超微结构
由图6可知,相同倍数下,改性前后SDF具有不同的超微结构。未改性SDF表面无缝隙,结构致密,呈现团状结构;改性SDF表面有蜂窝状孔隙,凹凸不平,比表面积增大,组织被颗粒化,且形状不规则[18]。表明高压-超声波协同法改性SDF聚合度较低,可能导致两者的理化性质如溶胀性和吸水性的不同。

注:1为未改性SDF(×3 000);2为未改性SDF(×500);3为改性SDF(×3 000);4为改性SDF(×500)
2.3.2 高压-超声波协同法改性前后SDF的X射线衍射结果
由图7可知,未改性SDF和改性SDF在扫描角度2θ为21°~24°均有明显的结晶衍射峰,表明它们具有纤维素I型的特征[19-20],其中,改性SDF的衍射强度低于未改性SDF,表明高压-超声波协同法使结晶区纤维素分子间的氢键破坏,纤维素分子发生部分降解,但并没有使SDF的晶型发生改变。
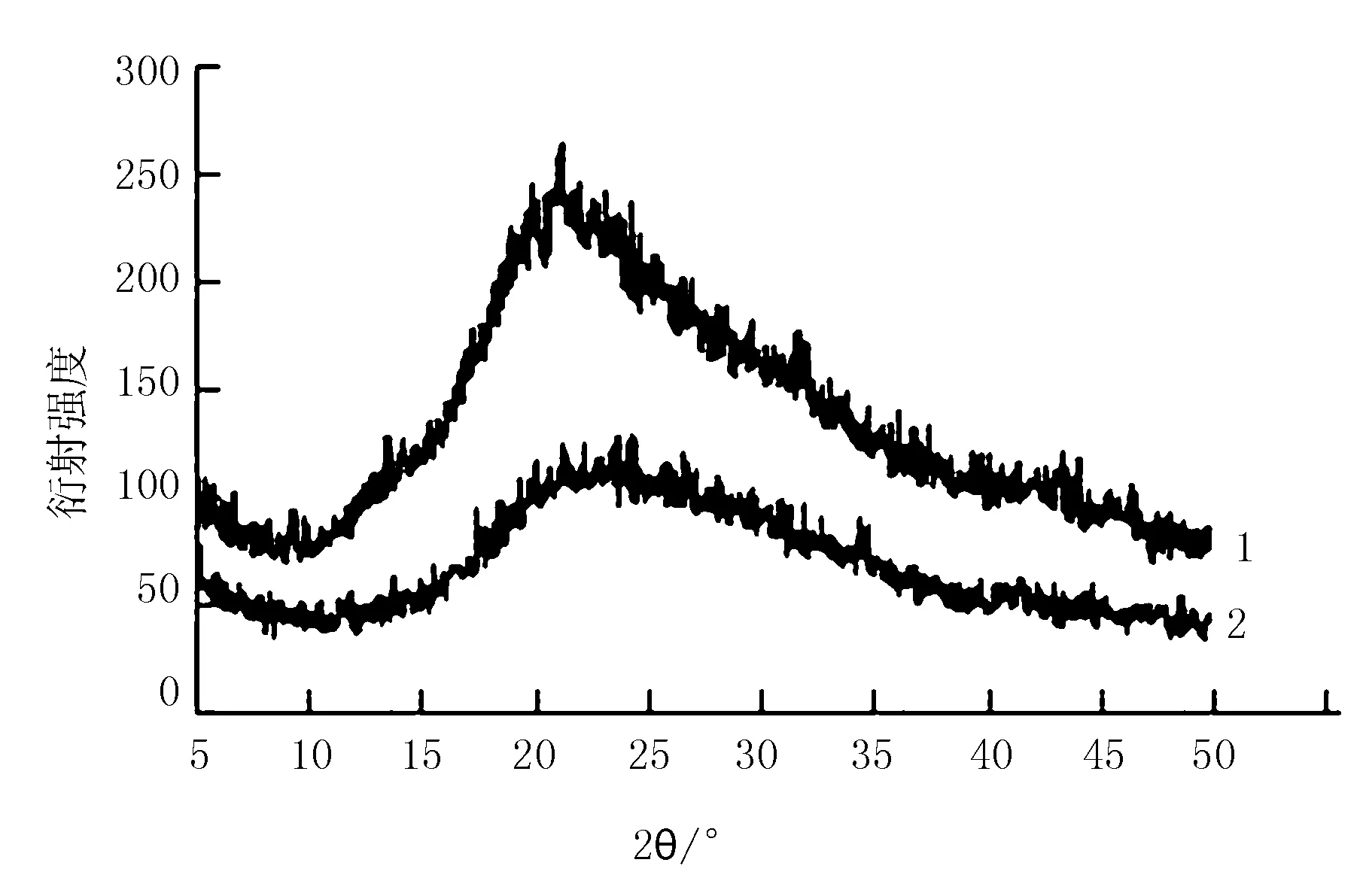
注:1为未改性SDF;2为改性SDF。
2.3.3 高压-超声波协同法改性前后SDF傅里叶变换红外光谱
高压-超声波协同法改性前后SDF的红外光谱见图8。
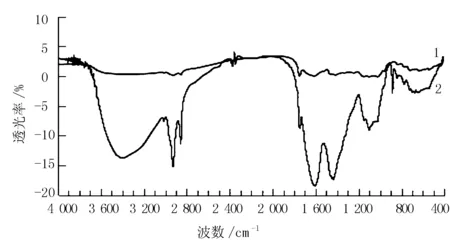
注:1.为未改性SDF;2.为改性SDF
由图8可知,改性SDF在3 398.57 cm-1出现的宽吸收峰是由于O-H伸缩振动产生,可能是高压-超声波协同法破坏了IDF中的糖苷键,暴露出更多O-H[19];改性前后的SDF均在2 924.09 cm-1出现的尖峰是由C-H振动引起,但改性SDF的峰强度更大,1 745.58 cm-1的尖峰是由CHO的碳氧键的收缩振动形成,1 200.00 cm-1附近的宽吸收峰,也是半纤维素糖单元中C-O-C的特征吸收峰,880~890.00 cm-1附近的吸收峰为糖单元间β-糖苷键的特征吸收峰,改性SDF的峰强度明显高于改性前,且未出现位移,表明高压-超声波协同法未对主要成分产生影响。
3 结论
以PIDF为试验材料,利用高压-超声波协同法进行改性。在固定条件压力0.1 MPa、温度121 ℃、时间20 min下,采用响应面法优化得到高压-超声波协同法的最佳改性条件为pH值12、超声波功率378 W、超声波时间92 min。验证试验结果与回归模型预测的理论值接近,RSD小于2%,表明采用响应面法优化的条件具有真实可靠性。利用扫描电子显微镜、X射线衍射和傅里叶变换红外光谱将改性前后SDF的结构进行比较的结果表明,未改性SDF结构致密,表面光滑,高压-超声波协同法改性SDF的表面疏松多孔且结晶束较少,且主要成分未发生明显改变,改性效果良好。
