选择性肝孤儿受体激动剂5α,6α-环氧胆甾醇前药的合成
2019-08-28徐孝浪郭晓强张梦晗李小红王虹宇何源超
徐孝浪, 郭晓强, 张梦晗, 李小红,王虹宇, 周 闯, 何源超
(1. 成都大学 药学与生物工程学院, 四川 成都 610106; 2. 成都大学 药食同源植物资源开发四川省高校重点实验室, 四川 成都 610106;3. 成都大学 四川抗菌素工业研究所, 四川 成都 610051)
肝孤儿受体(LXRs)信号传导是一个调控胆固醇代谢、运输的途径[1~2],LXRs是一种配体依赖型核受体,有LXRα和LXRβ两种亚型,研究发现它可能成为治疗高胆固醇血症、动脉粥样硬化以及预防心脑血管疾病的新作用靶点[3~7].在LXRs配体的研究中,氧化型固醇24S、25环氧胆固醇是较早发现的LXRs非选择性激动剂,LXRα和LXRβ两种亚型都会与之结合.此外脑甾醇、22R-羟基胆固醇等氧固醇都具有LXRs激动作用[8],然而动物实验表明,LXRs激动剂会导致肝脏脂肪堆积以及使血液中甘油三酯的水平异常上升,这些副作用与LXRs诱导SREBP1c表达有关[9],因此,开发高效的LXRs选择性激动剂是一个亟待解决的问题[10].近年来研究[11]发现5α,6α-环氧胆甾醇(2a)在一些LXRs应答基因中表现出激动作用,并且在与LXRs的亲和力测试中发现2a具有比24S、25环氧胆固醇更好的亲和力;除此之外,2a与LXRβ型受体不会结合,与LXRα型受体结合度较高(IC50=0.08 μmol/L;EC50=1.7 μmol/L),因此2a有可能成为一种选择性的LXRs激动剂.但是2a本身是一个甾体类化合物,脂溶性很大,而水溶性较差,在一定程度上降低了2a在机体内的生物利用度.本文以2a为先导化合物,对C-3位羟基进行化学结构修饰,首先胆甾醇经m-CPBA(a)处理得到关键中间体2a,2a分别与戊二酸酐、顺丁烯二酸酐和烟酸在DCC/DMAP(b)条件下进行酯化反应制得酯化衍生物2a-1、2a-2、2a-3;另外2a分别与2,3,4,6-O-苯甲酰基-D-溴代葡萄糖、2,3,4,6-O-苯甲酰基-D-溴代半乳糖和2,3,4-O-苯甲酰基-L-溴代阿拉伯糖在Ag2CO3催化下进行糖苷化反应,最后经CH3ONa脱保护(c)制备糖苷化衍生物2s-1、2s-2、2s-3(如图1所示),目标化合物均通过1H NMR、13C NMR和MS确证其结构,以期能得到2a具有合适脂水分配系数的前药,为2a的结构修饰提供一个可行的路径.
1 实验部分
1.1 试剂胆固醇,生物纯;二环己基碳二亚胺(DCC),分析纯;4A分子筛、强酸型阳离子交换树脂,成都市科隆化学品有限公司;间氯过氧苯甲酸(m-CPBA)、4-二甲氨基吡啶(DMAP)、戊二酸酐、顺丁烯二酸酐、烟酸、D-葡萄糖、D-半乳糖、L-阿拉伯糖、碳酸银、甲醇钠、二氯甲烷、石油醚、乙酸乙酯、四氢呋喃,均为分析纯,上海泰坦科技股份有限公司.
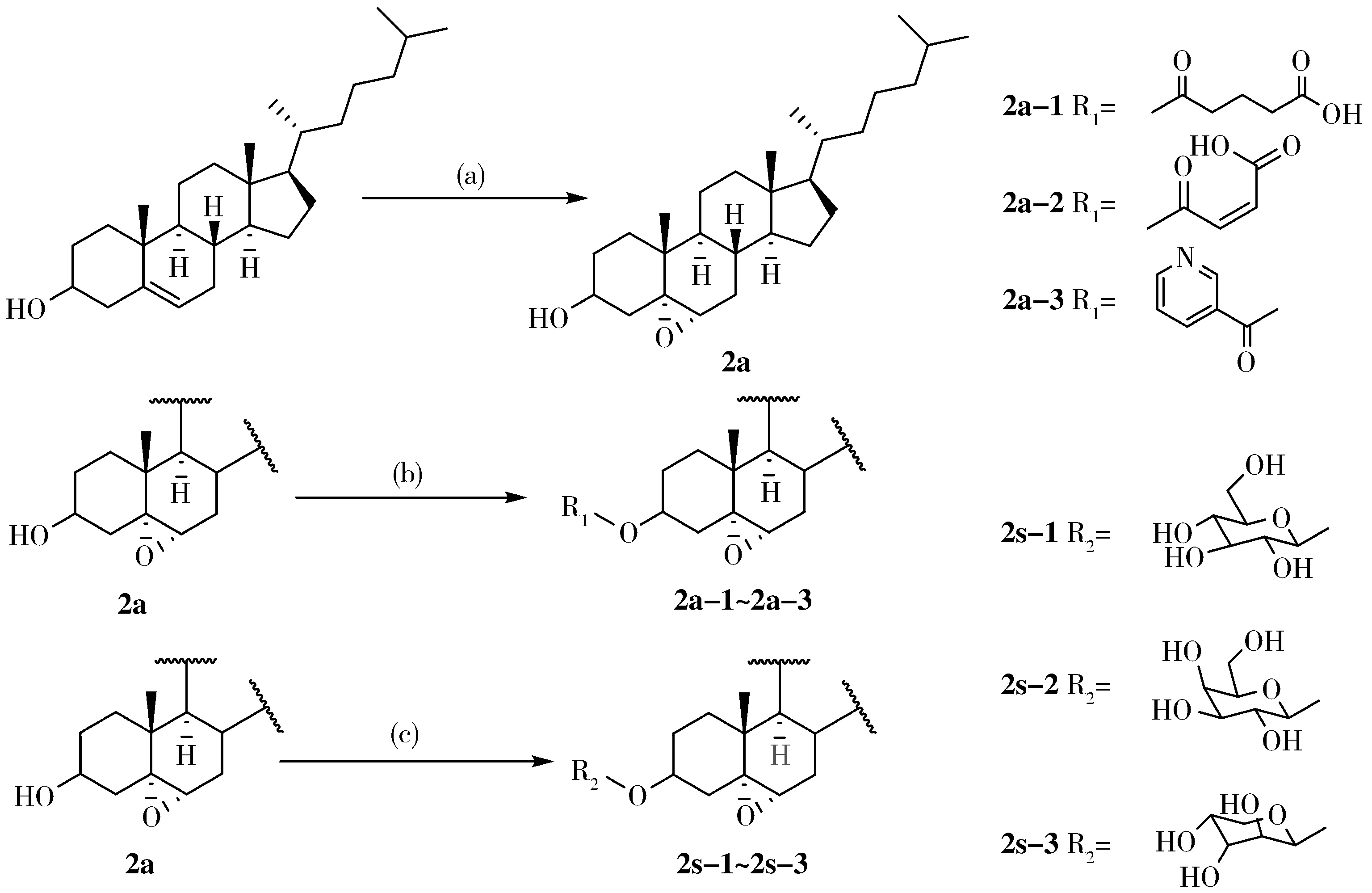
图 1 5α,6α-环氧胆甾醇衍生物合成路线
Fig.1Syntheitcrouteof5α,6α-epoxycholesterolderivatives
1.2 仪器Avance III 400 MHz核磁共振波谱仪(Bruker),6210 LC/MS TOF型质谱仪(Agilent Technologies),DTC-22型隔膜真空泵(东京理化器械株式会社),N-1100V-WD型旋转蒸发仪(东京理化器械株式会社),PSL-2000型恒温磁力搅拌水槽(东京理化器械株式会社),DHG-9246A电热恒温鼓风干燥箱(上海精宏试验设备有限公司),SHB-Ⅲ型循环水式多用真空泵(郑州长城科工贸有限公司),ZF-20D型暗箱式紫外分析仪(上海宝山顾村电光仪器厂),MP300型全自动熔点仪(济南海能仪器股份有限公司).
1.3 合成
1.3.12a的合成 50 mL单口瓶中加入胆固醇(0.5 g,1.29 mmol)和10 mL二氯甲烷,搅拌使胆固醇溶解,分批加入m-CPBA(0.25 g,1.55 mmol),室温搅拌反应18 h(TLC检测,磷钼酸显色;V(石油醚)∶V(乙酸乙酯)=2∶1).所得有机相在分液漏斗中依次用20 mL饱和亚硫酸钠溶液、20 mL饱和硫代硫酸钠溶液和50 mL饱和氯化钠溶液洗涤,分出有机相,无水硫酸镁干燥,减压蒸去二氯甲烷得到白色固体,经柱层析[洗脱剂:V(石油醚)∶V(乙酸乙酯)=10∶1]纯化得白色粉末0.45 g,收率85%.m.p∶141~142 ℃.1H NMR(400 MHz,CDCl3)δ:3.90(ddd,J=16.1,11.2,4.8 Hz,1H),2.92(d,J=4.4 Hz,1H),1.06(s,3H),0.90(s,3H),0.87(d,J=1.9 Hz,3H),0.86(d,J=1.8 Hz,3H),0.61(s,3H).13C NMR(101 MHz,CDCl3)δ:68.6、65.9、59.4、56.8、55.8、42.5、42.3、39.7、39.4、36.1、35.7、34.8、32.4、30.9、29.8、28.8、28.0、24.0、23.8、22.8、22.5、20.6、18.6、15.9、11.8.ESI-MSm/z:425.34[M+Na]+.
1.3.22a-1的合成 50 mL单口瓶中加入2a(0.05 g,0.12 mmol)和戊二酸酐(0.014 g,0.12 mmol)、四氢呋喃5 mL,剧烈搅拌下,依次加入DCC(0.05 g,0.24 mmol)和DMAP(0.03 g,0.24 mmol).升温至40 ℃反应48 h(TLC检测,磷钼酸显色;V(石油醚)∶V(乙酸乙酯)=2∶1).所得反应液减压蒸干用20 mL乙酸乙酯溶解,水洗分出有机相,1 mol/L盐酸洗涤,无水硫酸镁干燥,减压蒸馏得到的无色透明油状液体经柱层析[洗脱剂:V(二氯甲烷)∶V(甲醇)=10∶1]纯化得白色固体0.058 g,收率90%.m.p∶131~133 ℃.1H NMR(400 MHz,CDCl3)δ:4.98~4.92(m,1H),2.89(d,J=4.4 Hz,1H),2.41(t,J=7.3 Hz,2H),2.34(t,J=7.3 Hz,2H),1.06(s,3H),0.87(d,J=6.5 Hz,3H),0.85(d,J=3.0 Hz,3H),0.84(d,J=3.2 Hz,3H),0.59(s,3H).13C NMR(101 MHz,CDCl3)δ:178.7、172.0、71.5、65.4、59.2、56.7、55.8、42.3、39.4、36.1、35.7、35.0、33.4、32.9、32.1、29.8、28.7、28.0、27.2、23.9、22.8、22.5、20.5、19.8、19.5、18.6、15.8、11.8.ESI-MSm/z:517.39[M+H]+.
1.3.32a-2的合成 参照2a-1的合成方法进行制备,顺丁烯二酸酐(0.014 g,0.14 mmol)、[洗脱剂:V(二氯甲烷)∶V(甲醇)=20∶1]纯化得白色固体0.055 g,收率88%.m.p∶129~131 ℃.1H NMR(400 MHz,CDCl3)δ:6.47(d,J=12.9 Hz,1H),6.32(d,J=12.9 Hz,1H),5.14(ddd,J=16.3,11.3,4.9 Hz,1H),2.94(d,J=4.4 Hz,1H),1.10(s,3H),0.89(d,J=6.5 Hz,3H),0.87(d,J=1.9 Hz,3H),0.85(d,J=1.9 Hz,3H),0.61(s,3H).13C NMR(101 MHz,CDCl3)δ:168.2、164.2、137.2、129.4、75.1、65.1、59.5、56.8、55.9、42.5、42.4、39.6、36.2、35.8、35.1、32.1、29.9、28.8、28.1、27.0、24.1、23.9、22.9、22.6、20.7、18.7、15.9、12.0.ESI-MSm/z:539.31[M+K]+.
1.3.42a-3的合成 参照2a-1的合成方法进行制备,烟酸(0.02 g,0.18 mmol)、[洗脱剂:V(石油醚)∶V(乙酸乙酯)=4∶1]纯化得白色粉末0.054 g,收率86%.m.p:146~148 ℃.1H NMR(400 MHz,CDCl3)δ:9.16(s,1H),8.72(s,1H),8.29(d,J=7.9 Hz,1H),7.39(s,1H),5.23-5.14(m,1H),2.88(d,J=4.3 Hz,1H),1.27(s,3H),1.07(s,3H),0.80(s,3H),0.79(s,3H),0.55(s,3H).13C NMR(101 MHz,CDCl3)δ:164.4、153.2、150.8、137.0、126.5、123.2、72.7、65.2、59.2、56.7、55.8、42.4、42.3、39.5、39.3、36.2、36.1、35.7、35.0、32.1、29.8、28.7、28.0、28.0、27.3、24.0、23.8、22.8、22.5、20.6、18.6、15.9、11.8.ESI-MSm/z:530.36[M+Na]+.
1.3.52s-1的合成 50 mL单口瓶中加入2a(0.20 g,0.49 mmol)和2,3,4,6-O-苯甲酰基-D-溴代葡萄糖(0.64 g,0.98 mmol)、Ag2CO3(0.13 g,0.49 mmol)、粉状分子筛200 mg、无水乙醚20 mL搅拌溶解,室温反应2.5 h,(TLC检测,磷钼酸显色;V(石油醚)∶V(乙酸乙酯)=2∶1).反应完全后,抽滤,所得滤液减压蒸馏后经快速柱层析[洗脱剂:V(石油醚)∶V(乙酸乙酯)=4∶1]纯化得白色固体0.46 g.所得白色固体用10 mL甲醇溶解,室温下加入5 mL质量分数30%甲醇钠甲醇溶液反应2 h,(TLC检测,磷钼酸显色;氯仿∶甲醇=5∶1)反应完毕后,加入15 mg强酸型阳离子交换树脂调pH=6~7,抽滤,减压蒸干,所得棕色固体经快速柱层析[洗脱剂:A=V(二氯甲烷)∶V(甲醇)=20∶1]纯化得白色固体粉末0.23 g,总收率82%.m.p∶224~225 ℃.1H NMR(400 MHz,CDCl3)δ:8.08(d,J=7.5 Hz,2H),7.98(d,J=7.5 Hz,2H),7.91(d,J=7.4 Hz,2H),7.79~7.74(m,2H),7.61(dd,J=14.5,7.2 Hz,2H),7.43(dt,J=17.4,5.4 Hz,8H),7.24(d,J=7.8 Hz,2H),6.03(d,J=5.2 Hz,1H),5.73(d,J=2.5 Hz,1H),5.47(d,J=8.8 Hz,1H),4.75~4.71(m,1H),4.47(dd,J=12.1,2.7 Hz,1H),4.33(dd,J=12.1,4.8 Hz,1H),4.05(d,J=4.1 Hz,1H),3.70~3.60(m,1H),2.85(d,J=4.3 Hz,1H),0.99(s,3H),0.86(d,J=2.7 Hz,3H),0.85(s,3H),0.84(d,J=1.8 Hz,3H),0.57(s,3H).13C NMR(101 MHz,CD3OD∶CDCl3=6∶1)δ:101.0、76.5、75.9、73.5、70.1、66.1、61.7、59.9、56.8、55.8、42.6、42.3、39.4、36.4、36.1、35.7、35.0、32.4、29.8、29.0、28.7、27.9、23.9、23.7、22.6、22.3、20.5、18.5、15.7、11.7.ESI-MSm/z:565.41[M+Na]+.
1.3.62s-2的合成 参照2s-1的合成方法进行制备,2,3,4,6-O-苯甲酰基-D-溴代半乳糖(0.64 g,0.98 mmol)、得白色固体粉末0.21 g,总收率74%.m.p∶222~224 ℃.1H NMR(400 MHz,CDCl3)δ:7.96~7.90(m,6H),7.65~7.61(m,2H),7.54(dd,J=10.0,4.2 Hz,3H),7.41~7.34(m,9H),6.18(d,J=5.0 Hz,1H),5.75(dd,J=3.9,2.3 Hz,1H),5.42(dd,J=6.2,4.1 Hz,1H),4.79~4.73(m,1H),4.59~4.54(m,1H),4.46~4.40(m,1H),4.37~4.31(m,1H),3.91~3.82(m,1H),2.85(d,J=4.3 Hz,1H),1.01(s,3H),0.88(s,3H),0.86(d,J=1.6 Hz,3H),0.85(d,J=1.9 Hz,3H),0.59(s,3H).13C NMR(101 MHz,V(MeOD)∶V(CDCl3)=6∶1)δ:93.5、67.3、66.7、65.3、62.8、60.4、57.7、52.7、51.3、48.7、47.6、34.4、33.9、31.1、31.0、27.8、27.5、27.3、26.5、24.0、21.4、20.5、20.3、19.4、15.4、15.2、13.6、13.4、12.1、9.6、6.7、2.8.ESI-MSm/z:565.41[M+Na]+.
1.3.72s-3的合成 参照2s-1的合成方法进行制备,2,3,4-O-苯甲酰基-L-溴代阿拉伯糖(0.51 g,0.98 mmol)、得白色固体粉末0.18 g,总收率78%.m.p∶209~211 ℃.1H NMR(400 MHz,CDCl3)δ:8.06(d,J=7.2 Hz,2H),7.99(d,J=7.2 Hz,2H),7.93(d,J=7.3 Hz,2H),7.57~7.53(m,2H),7.46~7.40(m,5H),7.31(d,J=7.7 Hz,2H),5.69~5.65(m,2H),5.55(dd,J=8.9,3.4 Hz,1H),4.81(d,J=6.4 Hz,1H),4.32(dd,J=12.9,3.9 Hz,1H),3.95~3.89(m,2H),3.87(s,1H),2.78(d,J=4.3 Hz,1H),0.98(s,3H),0.89(s,3H),0.86(d,J=1.8 Hz,3H),0.85(d,J=1.8 Hz,3H),0.59(s,3H).13C NMR(101 MHz,V(MeOD)∶V(CDCl3)=6∶1)δ:103.0、76.7、74.1、72.2、69.4、67.1、66.7、60.7、58.2、57.0、43.9、43.3、40.6、40.5、37.4、37.1、36.8、36.0、33.5、30.9、30.0、29.8、28.9、24.9、24.7、23.1、22.9、21.5、19.1、16.2、12.3.ESI-MSm/z:535.40[M+Na].
2 结果与讨论
2.1 酯化反应条件的探讨酯化反应实验中首先使用Py/DMAP体系制备2a-1~2a-3,反应结果均分离到2个主要产物,1H NMR鉴定其中一个为目标产物(收率约10%),而另外一个为2a骨架中环氧基开环后的酯化产物(收率约80%),分析原因可能是吡啶作为碱的条件下环氧基更易受酸酐/羧酸的进攻而开环,反应条件过于剧烈.随后采用温和的DDC缩合法,成功减少了环氧基开环副产物的生成,但是收率很低(30%~40%).为了提高产品收率,本文重点探究了DCC以及DMAP的投料摩尔比对2a酯化反应收率的影响,固定2a投料量为50 mg,改变DCC、DMAP投料摩尔比,优化反应收率.实验结果如图2所示,可以发现,当2a投料量固定时,反应温度为40 ℃,溶剂为四氢呋喃,反应时间为48 h,DDC和DMAP的投料摩尔比为1∶1时,反应收率得到大幅提升(80%~90%),后期继续增大DCC与DMAP的投料摩尔比,其反应收率不再增加.
2.2 糖苷化反应条件的探讨Koenigs-Knorrs反应是糖化学中经典的成苷方法,首先采用Ag2CO3作为催化剂进行糖苷化反应,但是在反应过程中发现其收率较低(30%左右),TLC观察到大量未反应完的原料以及分解的卤代糖.通过干燥溶剂、补加大量卤代糖、升高反应温度也无明显改善.经分析知:反应过程中生成了卤化氢和水,这2种因素很可能会阻碍糖苷键的形成.因此,在原反应条件下加入粉状4A分子筛,以除去生成的水并吸收卤化氢,同时将溶剂无水化处理,结果反应收率大为改善,由原来的30%左右提高到80%~95%.图3为不同的分子筛用量对反应收率的影响,结果表明,随分子筛用量增加,收率明显增加,但当分子筛用量达到200 mg后,反应收率变化不明显.
2.3 目标化合物结构确证本文所合成的目标化合物2a-1~2a-3、2s-1~2s-3均未见文献报道,1H NMR图谱中δ(H)=0.62,0.85,0.87,0.89,1.13左右为环氧胆甾醇骨架上的氢.2a-1图谱中δ(H)=2.34,2.41为戊二酸结构中2个亚甲基的质子峰;2a-2图谱中δ(H)=6.32,6.47为顺丁烯二酸结构中双键上的质子峰;2a-3谱图中δ(H)=7.38,8.29,8.76,9.20为烟酸结构中吡啶环上的质子峰;2s-1中δ(H)=3.15,3.26,3.35,3.68,3.85,4.32为葡萄糖上的氢质子信号,2s-2中δ(H)=3.46,3.50,3.74,3.84,4.28为D-半乳糖上的氢质子信号,2s-3中δ(H)=3.50,3.53,3.80,3.85,4.24为L-阿拉伯糖上氢质子信号.另外各目标化合物的13C NMR谱、质谱图也与各自的结构相符.
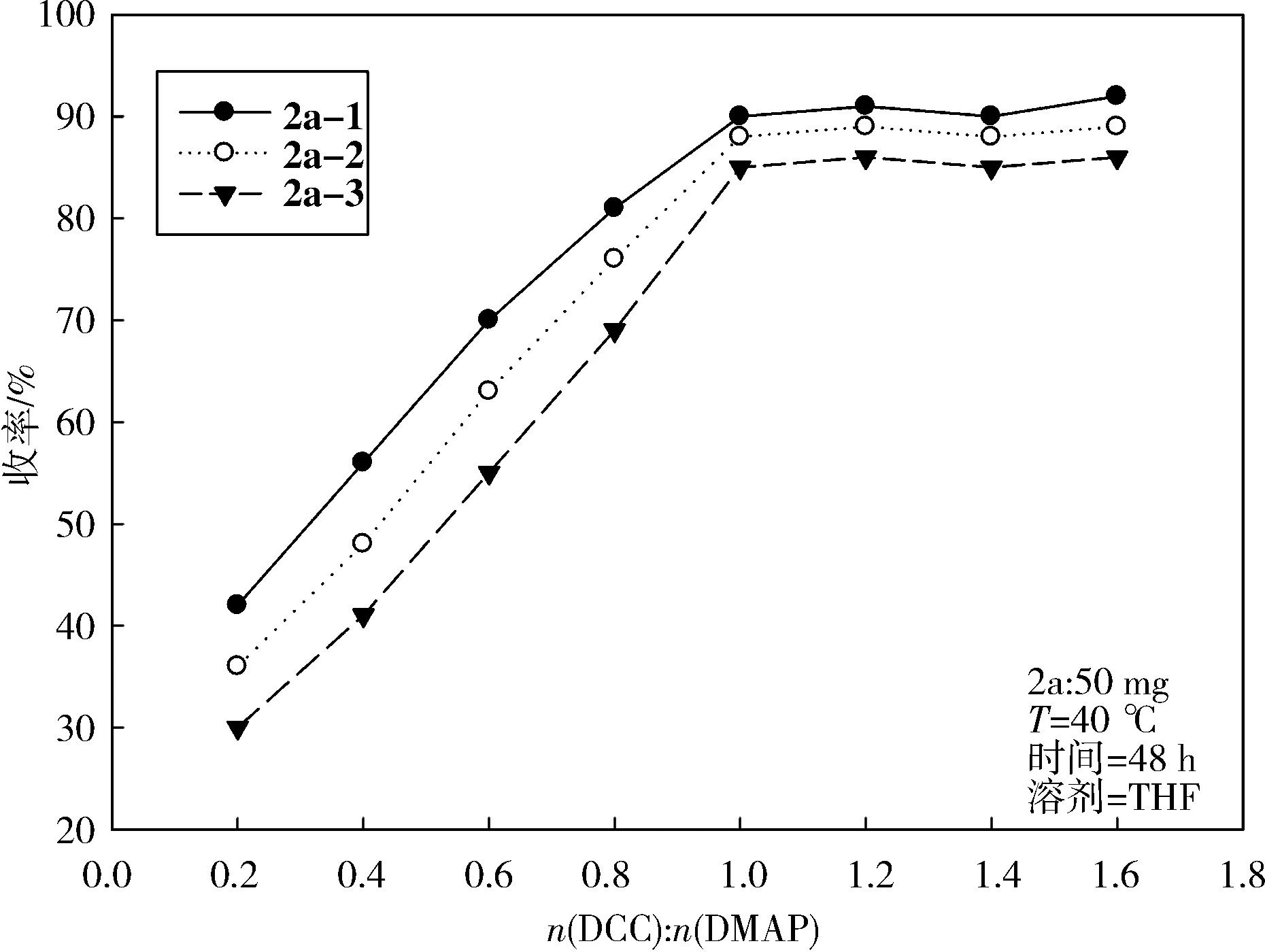
图 2 DCC和DMAP投料比(mole)对2a酯化反应收率的影响
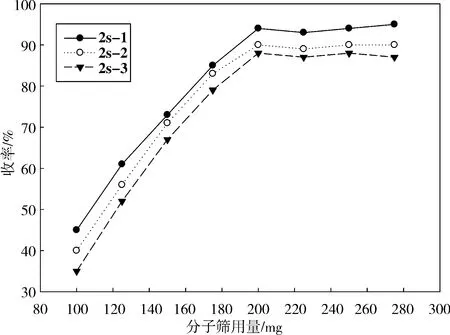
图 3 分子筛用量对2a糖苷化反应收率的影响
3 结束语
以调控胆固醇代谢、运输途径的LRXs为靶点,基于前药修饰策略,采用便宜易得的胆固醇为原料,通过有机过氧酸一步氧化得到2a,再分别与戊二酸酐、顺丁烯二酸酐和烟酸进行酯化反应制得2a酯化衍生物2a-1、2a-2、2a-3;另外2a分别与2,3,4,6-四-O-苯甲酰基-D-溴代葡萄糖、2,3,4,6-四-O-苯甲酰基-D-溴代半乳糖和2,3,4-四-O-苯甲酰基-L-溴代阿拉伯糖进行糖苷化反应制备糖苷化衍生物2s-1、2s-2、2s-3.成功探索出在环氧基不开环的前提下对其进行酯化、糖苷化修饰的路线,初步探索了缩合剂DCC/DMAP的投料摩尔比对目标化合物收率的影响,确定出2a酯化修饰线路中缩合剂的优选配比,大幅度提高了酯化反应收率.在糖苷化反应中加入粉状4A分子筛,及时移除阻碍糖苷形成的不利因素(H2O、HBr),糖苷化反应收率由约30%提高到90%左右.研究结果为以后针对2a展开更深入的结构修饰工作打下了一定的基础,也为2a的结构改造提供了有益的路径和策略参考.
