脱酚及pH偏移处理对菜籽蛋白体外模拟消化的影响
2019-08-28孙雪梅刘元法
孙雪梅,蒋 将,刘元法
(江南大学食品学院,食品科学与技术国家重点实验室,江苏无锡 214122)
油菜籽作为世界上第三大油料作物,在我国每年年产量均居第一。菜籽粕作为油脂加工的副产物,其蛋白含量较高,是一种丰富的植物蛋白资源。且菜籽蛋白具有平衡的氨基酸模式,含硫氨基酸的含量高于大豆蛋白[1]。但目前菜籽粕主要用作动物饲料或肥料,而限制菜籽蛋白在人类食品中应用的主要原因是存在抗营养因子,如植酸、硫苷、芥子酸、酚类物质及纤维等[2]。尽管引入了“双低菜籽”品种,但高含量的多酚和植酸依然是限制菜籽蛋白在食品中应用的重要因素[3]。多酚在碱性pH条件下,氧化形成醌类物质,与蛋白共价结合,从而导致蛋白溶液呈现墨绿色[4]。此外,多酚-蛋白复合物的形成还会降低菜籽粕的消化率[5]。因此,降低和脱除菜籽粕中的抗营养因子对提高菜籽蛋白的应用具有重要意义,同时脱酚后蛋白加工功能性有待进一步研究。
研究表明,通过一定的加工技术降低抗营养因子水平,可显著提高双低菜籽粕及其蛋白质在动物饲料中的消化率。其中,多酚对菜籽粕及蛋白的消化性质也起到重要作用。Qiao等[6]研究发现尽管菜籽中芥子酸对肉食鸡没有明显的毒性,但高含量下会影响氨基酸的消化率。Serraino等[7]的研究结果表明菜籽中酚酸的脱除能够增加氨基酸的释放速率。一方面,多酚可通过氢键、疏水相互作用及共价结合方式与蛋白形成复合体,从而降低蛋白的营养价值;另一方面,单宁也会阻碍消化酶的作用,影响蛋白水解[8]。
pH偏移作为一种有效的蛋白改性方法,通过将蛋白在强碱条件下维持一段时间后再将其调至中性,从而诱导球体蛋白进入“熔球态”,即蛋白质保持相对紧凑的结构(即保留大多数二级结构)但往往会失去一些三级结构的状态。处于“熔球态”中的蛋白,其溶解度、乳化性及抗氧化性质能够得到明显改善[9]。
本文采用体外模拟胃肠消化模型,通过水解度、蛋白电泳及微观结构等来研究脱酚及pH偏移处理对菜籽蛋白及蛋白乳液体系消化性的影响,从而为菜籽蛋白在食品领域的进一步开发与应用提供一定的实验依据。
1 材料与方法
1.1 材料与仪器
甘蓝型双低脱皮油菜籽 中国农业科学院油料作物研究所;大豆油 无锡欧尚超市;胃蛋白酶(3000 U/mg)、胰蛋白酶(2500 U/mg)、脂肪酶(100~500 U/mg) 上海源叶生物科技有限公司;氢氧化钠、氯化钠、磷酸二氢钠、磷酸氢二钠、三羟甲基氨基甲烷(Tris) 国药集团化学试剂有限公司;乙醇、正己烷等试剂 均为分析纯。
5804R离心机 德国Eppendorf公司;FE28 pH计 梅特勒公司;Nano Brook Omni多角度粒度与高灵敏度Zeta电位分析仪 美国布鲁克海文仪器公司;DM2700P偏光显微镜 德国徕卡公司。
1.2 实验方法
1.2.1 酚类化合物的脱除 将脱皮菜籽粉碎后,过80目筛,用正己烷-乙醇(10∶1,v/v)混合溶剂脱脂。根据Vuorela等[10]的方法对脱脂菜籽粉进行脱酚处理,略作修改。称取一定量的脱脂菜籽粕,以物料比1∶10 (w/v)加入有机试剂提取酚类化合物,鉴于食品安全要求,用70%乙醇(Et)室温下搅拌1 h,真空抽滤,将抽滤后的菜籽粕置于通风橱过夜,待提取蛋白。
1.2.2 菜籽分离蛋白的制备 根据Pirestani等[11]的方法提取菜籽分离蛋白,并作适当修改。称取一定量的脱酚菜籽粉,分散于去离子水中,料水比为1∶10 (w/v),用2 mol/L的NaOH调节pH至11.0,室温搅拌2 h后在8000×g,4 ℃离心30 min。用2 mol/L的HCl将上清液调至pH4.5,静置30 min,5000×g 离心20 min。将沉淀水洗3次,调节pH至7.0,冷冻干燥,得到蛋白Et,未脱酚处理提取的蛋白为C。参照GB 5009.5-2016,采用凯式定氮法测定蛋白质含量。
1.2.3 pH偏移处理 参照Jiang等[9]的方法,配制浓度为20 mg/mL的菜籽分离蛋白溶液。用2 mol/L NaOH调节pH至12.0,室温下搅拌1 h,随后用2 mol/L HCl再将其调回pH7.0,搅拌1 h。由C和Et处理后分别得到C12、Et12。对照组加入相应摩尔量的NaCl溶液。
1.2.4 蛋白体外模拟消化 参照Kaur等[12]的方法,将脱酚及pH偏移处理的蛋白溶液(6 mg/mL)用1 mol/L HCl调至pH2.0,加入胃蛋白酶(酶/底物1∶20,溶于0.1 mol/L HCl),在37 ℃恒温搅拌1 h。反应结束后,立即用0.9 mol/L NaHCO3将蛋白水解产物调至pH5.3,随后用1 mol/L NaOH继续调节pH至7.0,加入胰蛋白酶(酶∶底物=1∶20,溶于磷酸盐缓冲溶液),在37 ℃恒温反应1 h。蛋白水解期间,分别收集0、30、60、90及120 min的蛋白水解样品,沸水浴5 min灭酶。将样品置于-18 ℃保存用于进一步分析。
1.2.4.1 水解度测定 采用OPA(邻苯二甲醛)法[13]测定蛋白水解度。将7.620 g硼砂和200 mg十二烷基硫酸钠(SDS)溶于150 mL去离子水中,使其完全溶解。称取160 mg OPA试剂溶于4 mL无水乙醇,并转移到上述溶液。将176 mg 1,4-二巯基苏糖醇(DTT)加入到上述溶液中,定容至200 mL。配制0.1 mg/mL的丝氨酸标准样品,取400 μL标准样品与待测水解样品分别与3 mL OPA试剂混匀,以去离子水作空白,准确反应2 min,在340 nm处测定吸光值。

其中:OD:吸光值;X:样品重量(g);P:样品中蛋白含量(%);0.9516:丝氨酸标准溶液中的丝氨酸-NH2(meqv/L,毫当量每升);0.1:样品体积转换为L,由凯式定氮法测得。
其中:α=1.00,β=0.40。
其中:h0:水解反应前样品相对原始底物的水解度值;h1:水解反应后样品相对原始底物的水解度值;htot=7.8。
1.2.4.2 消化率测定 通过测定水解产物的可溶性蛋白浓度表征蛋白消化率。取1 mL水解产物与1 mL三氯乙酸(TCA)溶液(20%,w/v)混合,10000×g离心10 min,收集上清液,并用10% TCA溶液适当稀释,于280 nm处测定吸光值,使样品吸光值在保持在标曲范围内。以牛血清蛋白做标曲,得线性回归方程为:
y=0.5869x+0.0008,R2=0.9994。
其中:y为吸光值,x为标品浓度(mg/mL)。
根据线性回归分析上清液中可溶性蛋白浓度。
其中:C:样品蛋白浓度(mg/mL);OD样品:水解后的上清液样品吸光值;0.5869,0.0008:由牛血清标曲所得的参数;n:样品稀释倍数。
1.2.4.3 聚丙烯酰胺凝胶电泳 根据Schägger等[14]的方法稍作修改,制备聚丙烯酰胺凝胶电泳的浓缩胶与分离胶浓度分别为4%和16%(w/v)。将水解样品溶解于上样缓冲液(25 mL 0.5 mol/L Tris,20 mL甘油,40 mL 10% SDS,15 mL水,pH6.8),使蛋白最终浓度为2 mg/mL,上样量为15 μL。电泳在恒定电压下进行:初始电压30 V,进入分离胶时电压为100 V。
1.2.4.4 ABTS+·清除率 参照Ma等[15]的方法测定ABTS+·清除率。将7 mmol/L ABTS储备液和2.45 mmol/L高硫酸钾(最终浓度)混合,在室温、避光条件下静置12~16 h,形成ABTS自由基工作液。将工作液用0.2 mol/L PBS(pH7.4)稀释,使其在734 nm处的吸光度值为0.70±0.02。取20 μL蛋白水解样品与1980 μL工作液混合反应10 min,在734 nm处测定吸光值。以Trolox做标准曲线,得线性回归方程为:
y=0.3844x+0.001,R2=0.9971。
其中:y为吸光值,x为标品浓度(mmol/L)。
根据线性回归分析,结果以Trolox当量(mmol/L)表示。
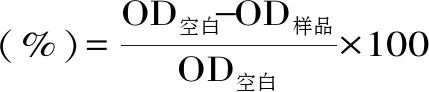
1.2.4.5 DPPH自由基清除率的测定 基于Zhou等[16]描述的方法。将1.5 mL的蛋白水解样品与等体积0.1 mmol/L DPPH溶液混合。振荡摇匀并在室温下反应30 min,在517 nm处测定吸光度。以Trolox做标准曲线,得线性回归方程为:
y=0.0233x+0.0216,R2=0.9985。
其中:y为吸光值,x为标品浓度(μmol/L)。
根据线性回归分析,结果以Trolox当量(μmol/L)表示。
1.2.5 蛋白乳液制备 将菜籽分离蛋白配制成浓度为10 mg/mL的蛋白溶液,与大豆油以1∶3 (v/v)的比例混合,用Ultra-Turrax高速分散机在13500 r/min的剪切速率下剪切2 min进行预乳化。然后将乳状液用AH-2010高压均质机在30 MPa压力下均质3次。
1.2.6 乳化性质的测定 将1.2.5中剪切过的预乳液迅速倒入25 mL小烧杯中,立即从距烧杯底部5 mm处取20 μL乳状液于试管中,在试管中加入5 mL 0.1%(w/v)SDS溶液,混匀后于500 nm处测定吸光度值A0;静置30 min重新取样测定吸光度值A30,以0.1% SDS溶液做空白对照。
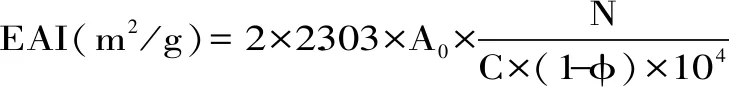
其中:N:稀释倍数;C:乳化液形成前蛋白溶液中蛋白质浓度(g/mL);φ:乳化液中油的体积分数(L/L);A0:0 min时乳液的吸光值;A30:30 min时乳液吸光值。
1.2.7 乳液模拟体外消化 乳液体外模拟消化参照Xu等[17]的方法略作修改。将40 mL乳液与等体积的模拟胃液(胃蛋白酶3.2 g,NaCl 2 g,HCl 7 mL,定容至1 L)混合,再用1.0 mol/L HCl溶液调至pH2.0,37 ℃恒温搅拌1 h,随后用2 mol/L NaOH调至pH7.0,终止反应。取出40 g水解产物,加入10 g模拟小肠液(脂肪酶1.6 mg/g,胰蛋白酶1.0 mg/g,胆盐10 mg/g,均为最终浓度),在37 ℃条件下继续搅拌1 h。反应结束后,沸水浴5 min灭酶。
1.2.7.1 微观结构 将乳液用去离子水稀释5倍,混匀,取一滴样品置于载玻片上,缓慢放置盖玻片,避免起泡,用DM2700P偏光显微镜的普通光学模式(物镜20×,目镜10×)下观察乳液油滴分布及聚集情况。
1.2.7.2 粒径测定 将乳液用去离子水稀释500倍,采用多角度粒度分析仪测定乳液粒径,分散相折射率为1.330,分散质折射率为1.492,测定温度为25 ℃。
1.3 数据处理
所有实验均重复2~3次,并且至少3次平行,结果表示为“平均值±标准偏差”形式。显著性分析采用SPSS 21.0通过Duncan法进行评估(p<0.05)。图表由Origin 8.0软件制作。
2 结果与分析
2.1 脱酚及pH偏移对蛋白体外消化的影响
2.1.1 水解度 蛋白水解度的变化通常用来解释蛋白消化率。脱酚及pH偏移处理的菜籽蛋白经过体外模拟胃肠消化后,消化产物的水解度变化如表1。随消化时间的延长,所有样品的蛋白水解度呈上升趋势。经过60 min胃蛋白酶水解后,C和C12的水解度相较于脱酚处理的样品(Et及Et12)分别提高了39.2%和68.4%,这表明蛋白经过脱酚处理后抑制了蛋白在胃消化阶段的水解。然而经过1 h(60~120 min)胰蛋白酶水解后,脱酚处理的样品(Et、Et12)水解度相较于空白(C、C12)分别增加了17.1%和22.9%,表明酚的脱除促进了蛋白在小肠阶段的水解。
可能的原因是脱酚处理促进了蛋白结构的展开,进一步诱导疏水聚集,掩盖了胃蛋白酶与蛋白的结合位点,从而使脱酚蛋白在胃蛋白酶消化阶段的水解较缓慢[18]。经过1 h的胃消化,诱导蛋白结构一定程度上被破坏。与空白相比,前期脱酚处理和pH偏移处理均使得蛋白结构更加松散,因此更有助于胰蛋白酶的消化。总的来说,菜籽蛋白中内源酚的存在会降低蛋白的消化率。此外,单纯的pH偏移处理对蛋白消化率无显著影响(p>0.05),但pH偏移处理结合脱酚处理可大大增加蛋白的消化率。
2.1.2 体外消化率 不同消化时间的蛋白消化产物通过TCA沉淀,其上清液中可溶性蛋白的浓度变化如图1。蛋白的原始浓度为6 mg/mL,未经蛋白酶消化前,TCA沉淀后的上清液蛋白浓度约为0.7 mg/mL,胃蛋白酶水解后,上清液中可溶性蛋白浓度显著增大到5 mg/mL左右。胃蛋白酶水解过程中(30~60 min),所有脱酚样品(Et和Et12)的可溶性蛋白浓度均低于其对应的空白样品,与水解度变化相同。仅pH偏移处理的样品(C12)与空白没有差异。即脱酚处理诱导的蛋白可溶性聚集阻碍了胃蛋白酶的水解,pH偏移处理对胃蛋白酶的水解无显著影响。而经过1 h的胰蛋白酶水解后,可溶性聚集被破坏,脱酚处理及胃蛋白酶的水解均促进了蛋白结构的展开,Et的可溶性蛋白浓度显著提高(p<0.05),比对照组提高了22.3%,这与蛋白水解度的变化趋势一致。同样的,pH偏移在胰蛋白酶水解阶段影响较小。
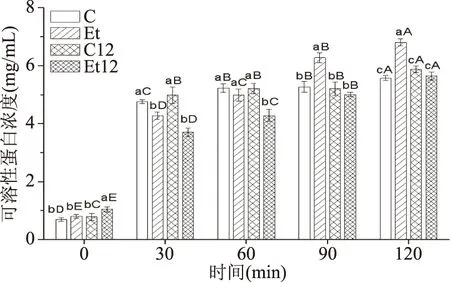
图1 脱酚及pH偏移处理对CPI体外模拟消化产物可溶性蛋白浓度的影响
2.1.3 SDS-PAGE 为了进一步考察体外模拟消化时蛋白酶对不同方式处理的蛋白亚基组成的影响,进行非还原SDS-PAGE分析(图2)。菜籽蛋白主要由12S(α和(亚基)和2S亚基组成,且2S(16 kDa)与12S(49 kDa)均由二硫键连接[19]。对于未经消化的天然CPI,脱酚处理可能会暴露更多的含硫氨基酸并促进二硫键的形成,因此,在电泳中表现出16 kDa条带的强度增加。经过胃蛋白酶水解后,所有亚基含量均减少,且在10 kDa处生成新的亚基。其中12S中(亚基在未脱酚组CPI(C和C12)明显减少,在脱酚组(Et和Et12)完全消失。经过胰蛋白酶消化,10 kDa亚基条带,未脱酚组浓度明显降低,脱酚组消失。与C相比,C12中2S和12S亚基含量均减少。但对于脱酚后样品,pH偏移处理对消化性无明显影响。总的来说,脱酚处理对蛋白消化性变化起到主导作用。这说明脱酚后蛋白结构发生一定程度展开,且原有的酚与蛋白的结合位点暴露出来,更有利于其与蛋白酶的结合。Duodu等[18]曾报道酚与蛋白结合形成复合物,并且酚类物质也具有一定的空间位阻效应,这都会阻碍酶对蛋白的作用。但脱酚结合pH偏移处理后蛋白展开程度较脱酚处理明显增大,且大量的疏水基团暴露诱导蛋白发生可溶性聚集[9],因此蛋白水解度显著增大(表1),但电泳结果没有显现出来,有可能是水解成较小分子量的肽,无法在电泳中显示。
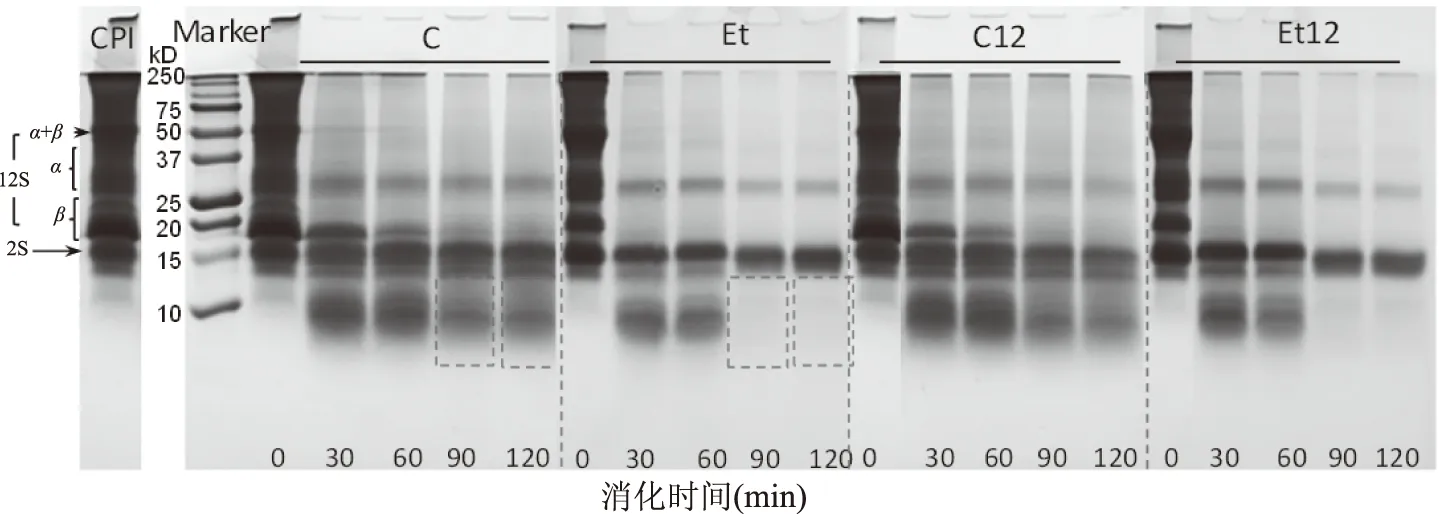
图2 脱酚及pH偏移处理对CPI体外模拟消化产物SDS-PAGE的影响
2.1.4 消化产物抗氧化性质 酚类物质一般具有较好的抗氧化性。蛋白自身也具有一定的抗氧化性,pH偏移处理后的蛋白由于结构展开,疏水基团暴露,抗氧化性明显增大[20]。脱酚及pH偏移处理后的蛋白体外消化产物的ABTS+·及DPPH自由基清除能力如图3所示。在整个消化过程中,ABTS+·及DPPH自由基清除能力均呈先增加后降低的趋势,且在胃蛋白酶水解60 min后达到最大值。类似的结果也曾被Zhou等[16]和Phongthai等[21]报道过。在图3A中,脱酚处理使得Et蛋白的ABTS+·清除能力降低了69.9%,然而pH偏移处理后的C12提高了172%,脱酚后的样品再经pH偏移处理,其抗氧化性大大增强(80.6%),这表明蛋白中酚的存在具有重要的抗氧化活性。同时,pH偏移促进了蛋白结构的部分展开,暴露了更多的氨基酸侧链基团及肽,提高了蛋白的自由基清除能力,弥补了脱酚后抗氧化性降低的缺陷。Jiang等[20]在豌豆蛋白中也发现了类似的结果。经过胃蛋白酶水解,消化产物的自由基清除能力均明显提高,其中C和C12,Et和Et12的ABTS+·及DPPH自由基清除能力分别相互接近,主要是由于胃蛋白酶的水解作用导致蛋白的水解及多肽的产生,使得大量的疏水氨基酸暴露出来。此外,在酸性条件下胃蛋白酶的水解导致一些结合酚的释放,增加了自由基清除能力[22]。经过1 h的胰蛋白酶的水解后,不同方式处理的蛋白消化产物的DPPH自由基清除能力降低了32%~62%,主要原因是胰蛋白酶选择性地切割碱性氨基酸(赖氨酸和精氨酸),消化产物极性增加使得与自由基反应更加困难[23]。
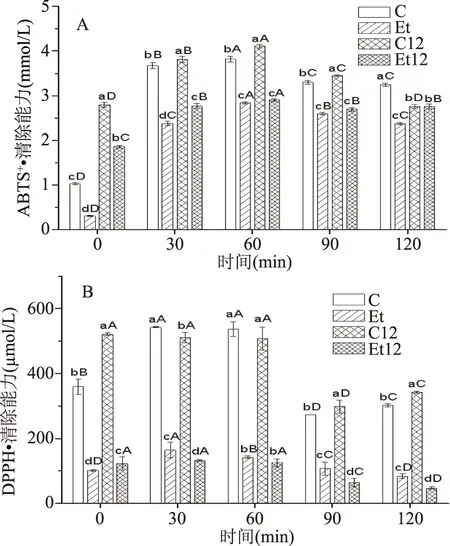
图3 脱酚及pH偏移处理对CPI体外模拟消化产物ABTS+·(A)和DPPH·(B)清除率的影响
2.2 模拟乳液体外消化
模拟乳液体外消化条件下的微观结构和粒径分布如图4和图5所示。图4表明,菜籽蛋白经过pH偏移后,乳化活性及乳化稳定性均有很大提高,因此选择以pH偏移处理后的蛋白为乳化剂来研究脱酚处理对乳液体外消化性质的影响。采用光学显微镜表征乳液在模拟体外消化过程中的微观结构变化(如图5)。在胃消化阶段所有乳液样品均处于相对稳定状态,经过30 min胃蛋白酶水解后,乳液聚集现象明显,出现乳液分层现象,被水解蛋白进入清液层,上层乳液中的蛋白是由部分水解蛋白和未水解蛋白组成。一方面,在pH2的胃蛋白酶消化过程中,蛋白表面所带电荷较pH7.5少,斥力减少,从而引起油滴聚集[24];另一方面,胃蛋白酶对油滴表面吸附的蛋白进行了水解,其作用位点是蛋白芳香族氨基酸和一些疏水残基[24],经过1 h胃蛋白酶消化,蛋白的三级四级结构已被基本破坏,蛋白的乳化能力降低从而诱导油滴聚集[25]。而Et12稳定的乳液液滴聚集程度较C12大,这是由于脱酚后蛋白结构较松散有助于蛋白酶的结合并发生水解,从而诱导乳液部分聚集。胃蛋白酶水解60 min后,乳液液滴变小,且分散较均匀,可能是由于随着水解时间的延长,大量多肽的形成,蛋白表面带电量增大,油滴的静电斥力增强。在小肠消化阶段,经过30 min的脂肪酶及胰蛋白酶的水解后,乳液呈现失稳现象,出现明显的浮油层。从显微镜结果观察,乳液相油滴分散更加均匀。在此阶段,由于胆盐的加入以及脂肪酶水解产生的游离脂肪酸会竞争性地取代油滴表面的蛋白[26],从而形成新的较稳定的乳液。脱酚处理后的蛋白乳液经过1 h的模拟小肠消化,油滴比对照组小。综合以上现象表明,蛋白经过脱酚处理后能够提高蛋白乳液的消化率。
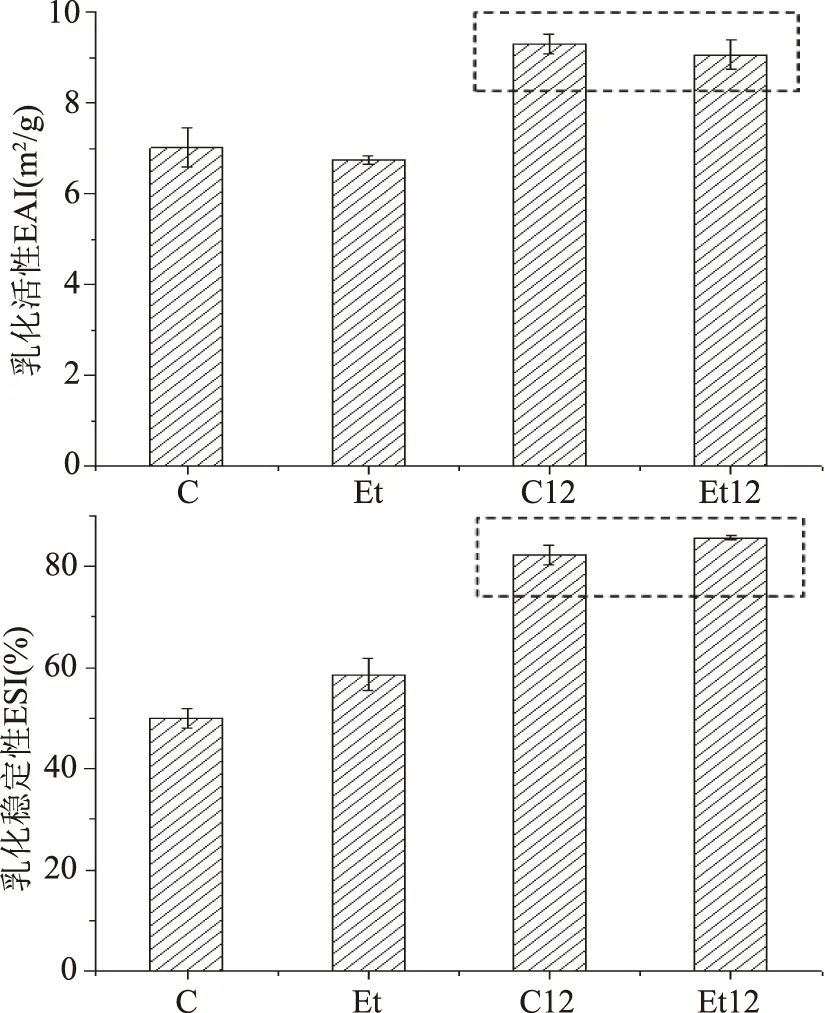
图4 脱酚和pH偏移处理的CPI乳液的乳化活性(EAI)和乳化稳定性(ESI)
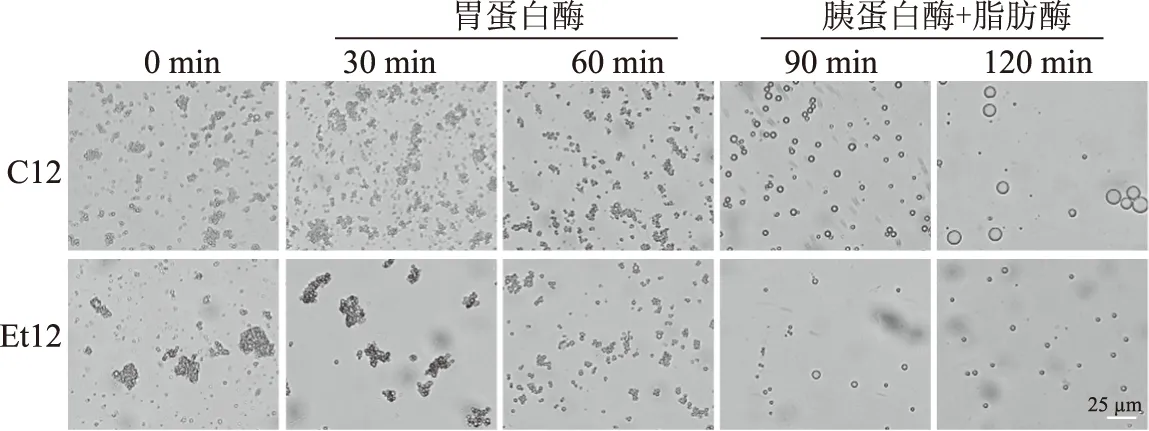
图5 CPI乳液体外模拟消化产物的微观结构图
为了进一步验证乳液在消化过程中的微观结构变化,对消化过程中乳液平均粒径进行测定(图6)。在胃蛋白酶水解30 min后,C12和Et12稳定的乳液平均粒径明显增大,分别增大了2.8和3.6倍,表明胃蛋白酶对界面蛋白的水解促进了液滴聚集,尤其是脱酚处理的蛋白乳液。60 min后,乳液粒径降至2.5~3.5 μm之间。经过1 h的胰蛋白酶及脂肪酶的水解后,C12稳定的乳液粒径相比在90 min时明显增大,且大于Et12稳定的乳液平均粒径,与图5中的乳液微观结构趋势一致。
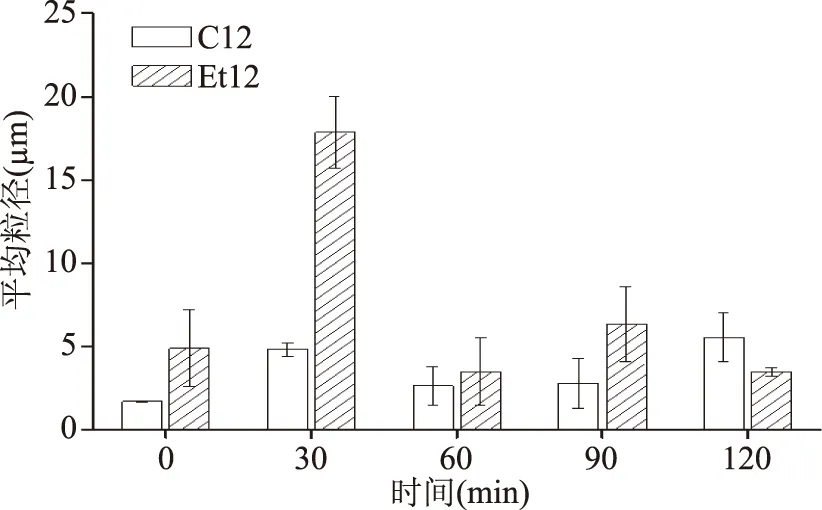
图6 脱酚处理对CPI乳液体外模拟消化产物的平均粒径的影响
3 结论
本实验针对脱酚处理及pH偏移改性的蛋白,及蛋白稳定的乳液来研究不同体系的体外消化。在模拟蛋白消化过程中,水解度及可溶性蛋白浓度变化的结果表明脱酚处理能显著提高蛋白的消化率,且脱酚处理结合pH偏移处理可最大程度提高蛋白体外消化率。SDS-PAGE的结果中小分子量条带的消失也证明了脱酚的效果。pH偏移对蛋白及消化产物的自由基清除能力有明显提高,并能改善由脱酚导致的较差的自由基清除能力,有望延长蛋白的保质期。乳液消化的研究结果表明,脱酚处理能促进乳液中蛋白的水解并降低油滴尺寸。因此,脱酚结合pH偏移处理可以为菜籽蛋白在人类食品中的应用提供理论指导。
