藤茶总黄酮固体脂质纳米粒处方的优化及其体外释药行为
2019-08-26杨毛毛罗花彩林珠灿郭素华
杨毛毛, 罗花彩, 沙 玫, 徐 伟, 林珠灿*, 郭素华*
(1. 福建中医药大学药学院, 福建 福州350122; 2. 福建中医药大学附属人民医院, 福建 福州350004)
藤茶为葡萄科蛇葡萄属植物显齿蛇葡萄Ampelopsis grossedentata (Hand-Mazz) W. T. Wang 的嫩茎叶[1], 具有清热解毒、 利湿消肿之功效, 主治黄疸型肝炎、 感冒风热、咽喉、 目赤肿痛等症[2]。 近年来研究表明, 藤茶中总黄酮含有量高达40%以上, 其中二氢杨梅素(又称福建茶素)为其特征性成分[3-4], 具有抗肿瘤、 抗炎抑菌、 抗氧化、 降糖降脂、 增强免疫功能、 保肝护肝、 抗动脉粥样硬化等多种活性[4-9], 但其在体内消除快、 口服吸收差、 生物利用度低[10-11], 严重影响了临床应用和体内药效。
固体脂质纳米粒是一种新型给药体系, 以固态的天然或合成的类脂(如卵磷脂、 三酰甘油等) 为载体, 将药物包裹或夹嵌于类脂核中, 制成粒径约为10~1 000 nm 的固体胶粒而成[12], 它是以生理相容性好、 体内可降解、 无生物毒性的脂质为基质, 既能增加药物稳定性, 又可延长半衰期, 进而提高药效成分生物利用度, 同时又具有控释作用和良好的靶向性, 以及纳米粒药物泄露少、 毒性低、 操作简单等优点[12-13], 可广泛应用于大规模生产, 但目前尚无藤茶总黄酮相关剂型处方与制备工艺的报道。 因此, 本实验制备藤茶总黄酮固体脂质纳米粒, 然后优化处方, 考察其体外释药行为, 为相关新剂型开发与临床前研究提供实验数据支撑。
1 材料
LC-20A 高效液相色谱仪、 SPD-M20A 紫外检测器(日本岛津公司); JY92-II 型超声波细胞粉碎机(宁波新芝生物科技股份有限公司); NICOMPTM380ZLS Zeta potential/particle sizer 纳米粒度仪(美国Waters 公司); DKZ 系列超级恒温水浴振荡器(上海一恒科技有限公司); UFC501096超滤管(10 kD, 美国Millipore 公司)。
二氢杨梅素对照品(批号160422, 上海源叶生物科技有限公司, 含有量≥98%); 杨梅苷(批号15081713, 含有量99.41%)、 杨梅素(批号15050810, 含有量98.31%)对照品(北京盈泽纳新化工技术研究院)。 藤茶总黄酮原料药由课题组自制(含有量≥80%)。 单硬脂酸油酯(批号20170213, 国药集团化学试剂有限公司); 泊洛沙姆188(批号WPAK527B, 德国BASF 公司)。 甲醇为色谱纯(德国默克公司); 其他试剂均为分析纯; 水为超纯水。
2 方法与结果
2.1 固体脂质纳米粒制备[14]采用熔融-超声法。 精密称取泊洛沙姆188 适量, 超纯水溶解, 加热至75 ℃恒温, 作为水相; 精密称取适量藤茶总黄酮原料药、 单硬脂酸甘油酯, 加少量乙醇与水相, 在同温状态下搅拌熔融, 作为油相, 待两相完全溶解且温度相同时, 将水相倒入油相中快速搅拌以充分混合, 混合液挥至无醇味时超声波细胞粉碎机(变幅杆调至6, 功率200 W, 每超声1 s 间隔2 s) 超声5 min, 即得, 室温冷却后于4 ℃冰箱中保存。 同法制备空白固体脂质纳米粒(不加藤茶总黄酮原料药)。
2.2 包封率、 载药量测定
2.2.1 色谱条件[15]TOP ODS-AQ 色谱柱 (4.6 mm×250 mm, 5 μm); 流动相甲醇(A) -0.1% 磷酸(B), 梯度洗脱(0 ~10 min, 35% A; 10 ~20 min, 35% ~80% A;20~30 min, 80% A); 检测波长252 nm (杨梅苷、 杨梅素)、 291 nm (二氢杨梅素); 体积流量1.0 mL/min; 柱温30 ℃; 进样量10 μL。 色谱图见图1。
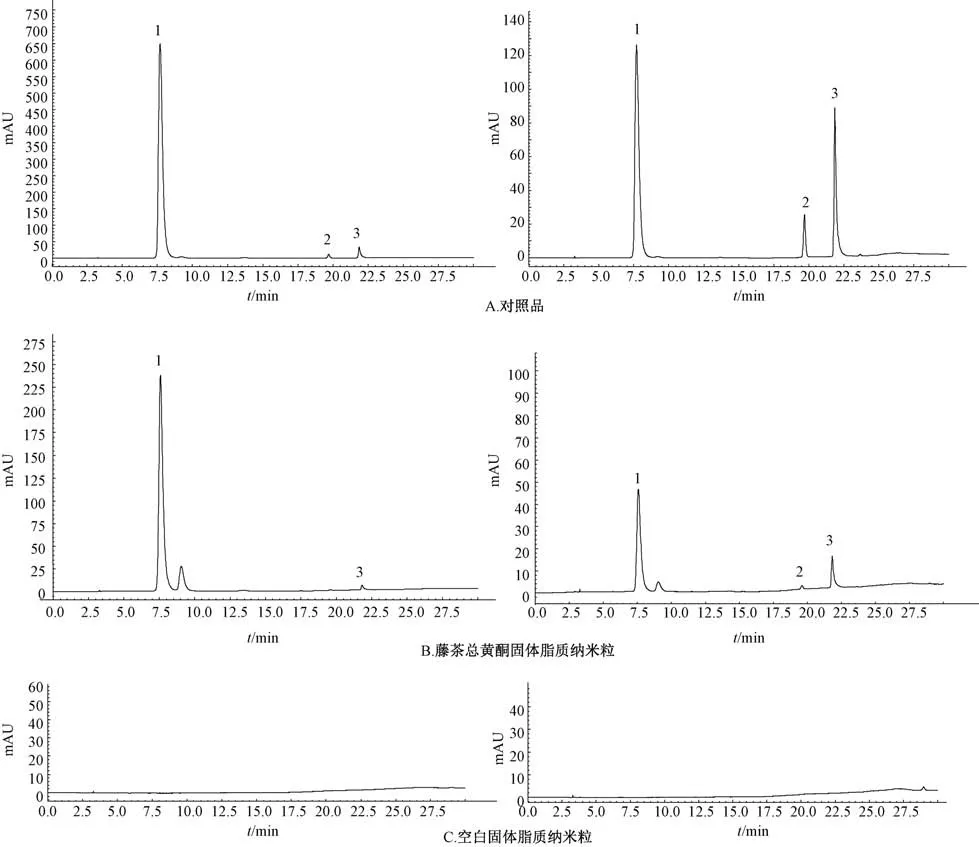
图1 各成分HPLC 色谱图
2.2.2 线性关系考察 精密称取二氢杨梅素、 杨梅苷、 杨梅素对照品适量, 置于10 mL 量瓶中, 甲醇定容, 即得对照品溶液(质量浓度分别为505.68、 6.29、 13.42 μg/mL),4 ℃下 保 存 备 用, 精 密 吸 取0.2、 0.5、 1.0、 2.0、 4.0、8.0、 10.0 mL 于10 mL 量瓶中, 甲醇稀释成系列质量浓度,在“2.2.1” 项色谱条件下进样测定。 以峰面积为纵坐标(Y), 溶液质量浓度为横坐标(X) 进行回归, 结果见表1,可知各成分在各自范围内线性关系良好。
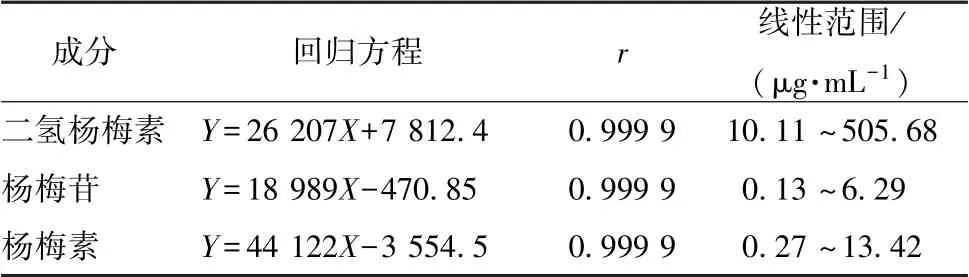
表1 各成分线性关系
2.2.3 精密度试验 精密吸取对照品溶液适量, 在“2.2.1” 项色谱条件下进样测定, 测得二氢杨梅素、 杨梅苷、 杨梅素日内精密度RSD 分别为0.62%、 0.82%、0.63%, 日间精密度RSD 分别为0.85%、 1.85%、 0.29%,表明该方法精密度良好。
2.2.4 稳定性试验 于0、 2、 4、 6、 8、 12、 24、 48、 72 h吸取同一供试品溶液, 在“2.2.1” 项色谱条件下进样测定, 测得二氢杨梅素、 杨梅苷、 杨梅素含有量RSD 分别为1.54%、 0.02%、 0.59%, 表明供试品溶液在72 h 内稳定性良好。
2.2.5 加样回收率试验 取各成分含有量已知(二氢杨梅素512.96 μg/mL、 杨梅苷6.39 μg/mL、 杨梅素13.76 μg/mL)的供试品溶液, 在“2.2.1” 项色谱条件下进样测定, 测得二氢杨梅素、 杨梅苷、 杨梅素平均加样回收率分别为101.44%、 101.68%、 102.53%, RSD 分 别 为 1.30%、2.56%、 2.34%。
2.2.6 测定方法
2.2.6.1 游离总黄酮 精密吸取固体脂质纳米粒混悬液500 μL 于超滤离心管中, 8 000 r/min 离心10 min, 取100 μL离心液, 甲醇定容至1 mL, 在“2.2.1” 项色谱条件下进样测定。
2.2.6.2 总黄酮总量 精密吸取固体脂质纳米粒混悬液500 μL 于10 mL 量瓶中, 甲醇加热溶解, 定容, 摇匀,0.45 μm 微孔滤膜过滤, 在“2.2.1” 项色谱条件下进样测定。
2.2.6.3 计算公式 包封率= [(W总-W游离) /W总] ×100%, 载药量= (W总-W游离) / (W脂质+W总-W游离) (W总表示总黄酮总含有量, W游离表示游离总黄酮含有量, W脂质表示单硬脂酸甘油酯质量)。
2.2.7 超滤回收率试验 精密吸取“2.1” 项下空白固体脂质纳米粒混悬液0.1、 0.3、 0.4 mL 各3 份于超滤离心管中, 分别精密加入0.4、 0.2、 0.1 mL 对照品 溶液,8 000 r/min离心10 min, 取滤液, 在“2.2.1” 项色谱条件下进样测定, 测得二氢杨梅素、 杨梅苷、 杨梅素超滤回收率分 别 为101.46%、 102.00%、 100.85%, RSD 分 别 为1.89%, 1.62%, 2.72%。
2.3 粒径测定 精密吸取“2.1” 项下固体脂质纳米粒混悬液100 μL, 超纯水稀释至10 mL 量瓶中, 摇匀, 纳米粒度仪测定其粒径和Zeta 电位。
2.4 处方优化 在前期预实验基础上, 以粒径(Y1)、 包封率(Y2)、 载药量(Y3) 为评价指标, 投药量(A)、 药脂比(B)、 乳化剂(泊洛沙姆188) 用量(C) 为影响因素,设计3 个水平进行处方优化。 因素水平见表2, 结果见表3。

表2 因素水平
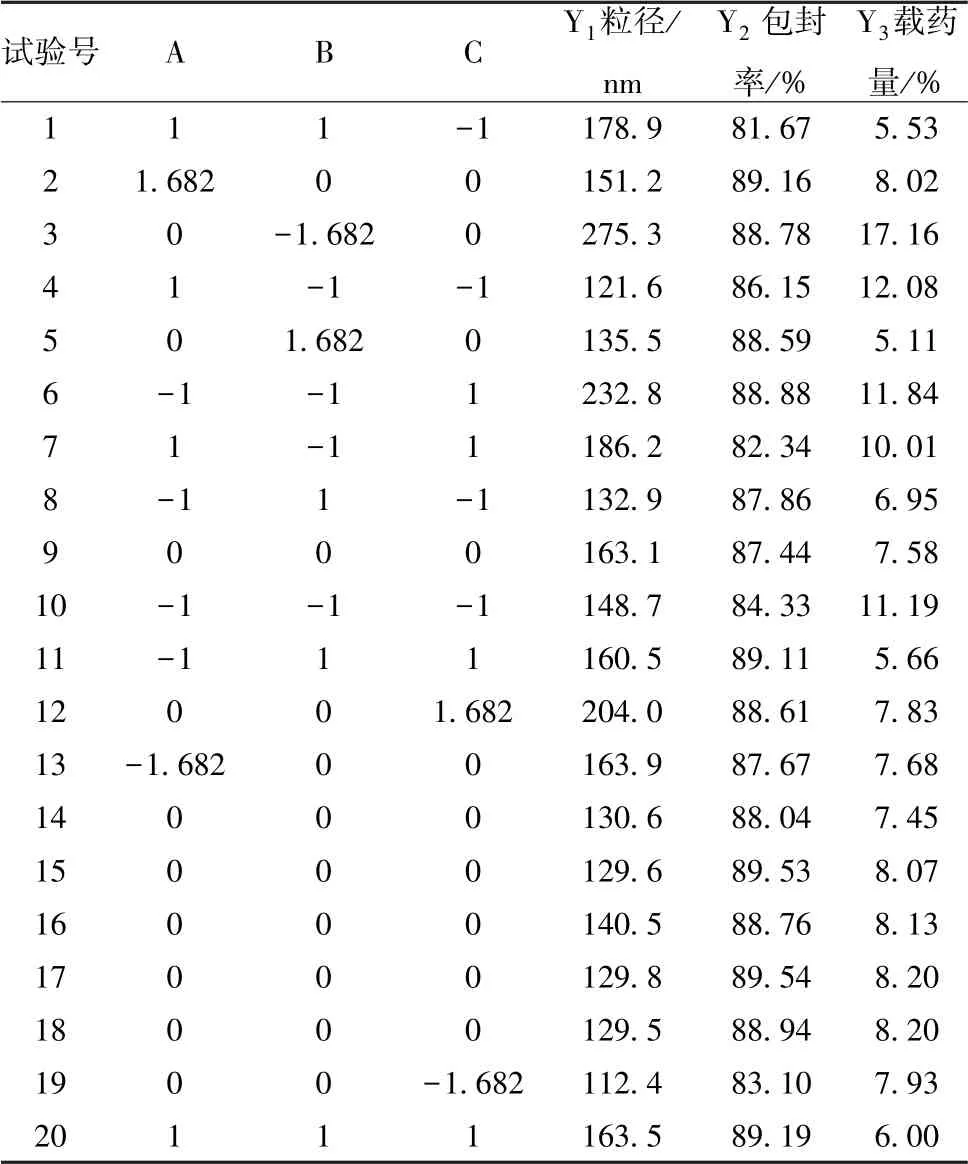
表3 试验设计与结果
通过Design-Expert 8.0 软件对表3 数据进行二项拟合,得到方程分别为Y1= 137.52-3.37A-21.13B+23.06C+15.34AB-7.81AC-17.06BC+5.03A2+21.95B2+5.26C2(r=0.906 5, P<0.05)、 Y2=87.38-0.61A+0.43B+1.38C (r=0.746 4, P >0.05)、 Y3= 7.95-0.11A-3.02B-0.18C-0.017AB-0.12AC+0.074BC-0.11A2+1.06B2-0.096C2(r=0.975 8, P<0.05), 可知各响应值与各因素水平的非线性拟合效果较好, 其中Y1、 Y3相关系数较大, 对粒径、 载药量的影响较显著(P<0.05)。
响应面分析见图2。 综合考虑后期大规模生产的可行性及经济成本, 确定最优处方工艺为投药量0.48%, 药脂比1 ∶5, 乳化剂用量6.6%, 粒径158.4 nm, 包封率86.13%,载药量12.15%。然后, 平行制备6 份固体脂质纳米粒混悬液, 测定粒径、包封率、 载药量, 结果见表4, 可知优化工艺重复性良好,具有可行性。 粒径分布、 Zeta 电位分别见图3~4。
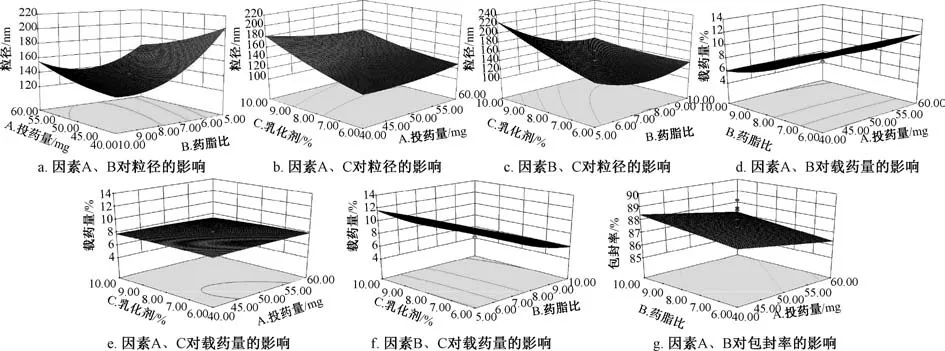
图2 各因素响应面图
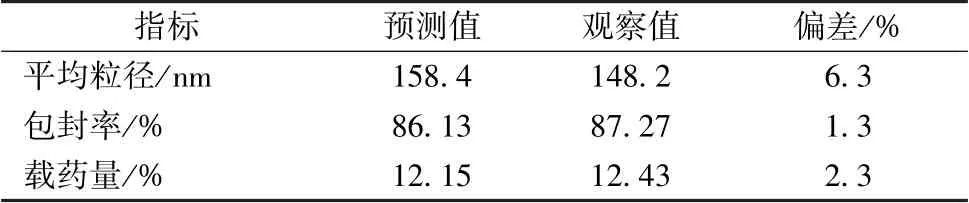
表4 验证试验结果(n=6)
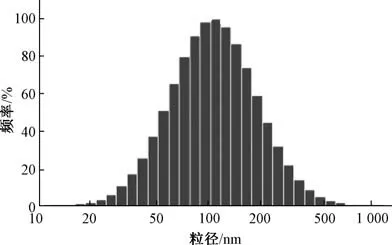
图3 纳米粒粒径分布
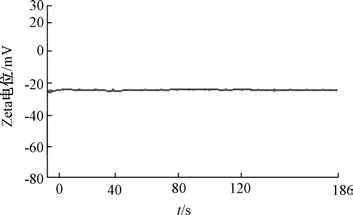
图4 纳米粒Zeta 电位
2.5 体外释药行为评价 采用平衡透析法[11], 释放介质为pH 5.8 磷酸盐缓冲液(含2%吐温80), 介质体积100 mL,介质温度(37±0.5)℃, 转速100 r/min。 精密移取适量固体脂质纳米粒混悬液(藤茶总黄酮含有量4.8 mg/mL) 于预先处理的透析袋(截留分子量8 000~14 000 Da) 中, 透析袋夹夹紧两端袋口后置于释放介质中, 于0.5、 1、 2、 4、8、 12、 24、 48 h 用100 mL 等温新鲜释放介质置换, 吸取1 mL释放介质, 0.45 μm 微孔滤膜过滤后弃去初滤液,HPLC 法测定二氢杨梅素、 杨梅苷、 杨梅素含有量; 精密称取原料药适量, 磷酸盐缓冲液(pH=5.8) 加热溶解, 制成质量浓度与固体脂质纳米粒相同的溶液, 同法进行释放试验, 计算累积释放率(Qi), 公式为Qi= (C1+C2+……+Ci) V/C0V0(V0、 C0分别为透析袋中加入样品的体积、 质量浓度, V 为透析介质体积, Ci为每个时间点测得的总黄酮质量浓度), 结果见图5。 由图可知, 原料药4 h 内累积释放率为97.59%, 基本释放完全; 纳米粒在4 h 前释放较快, 累积释放率为79.64%, 后期呈现缓释, 24 h 基本释放完全, 累积释放率为98.81%。
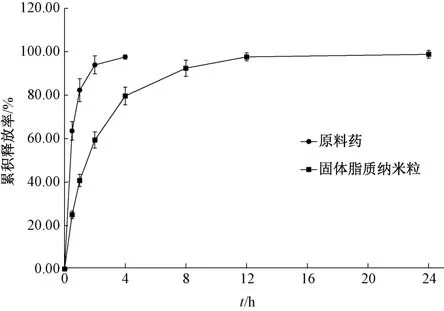
图5 样品体外释药曲线
在此基础上, 对固体脂质纳米粒、 原料药的体外释药数据进行零级、 一级、 Higuchi、 Weibull、 Korsmeyer-Peppas 模型的拟合, 结果见表5。 由表可知, 两者的释放动力学相同,Weibull 模型拟合效果最好(R2分别为0.999 5、 0.999 6)。
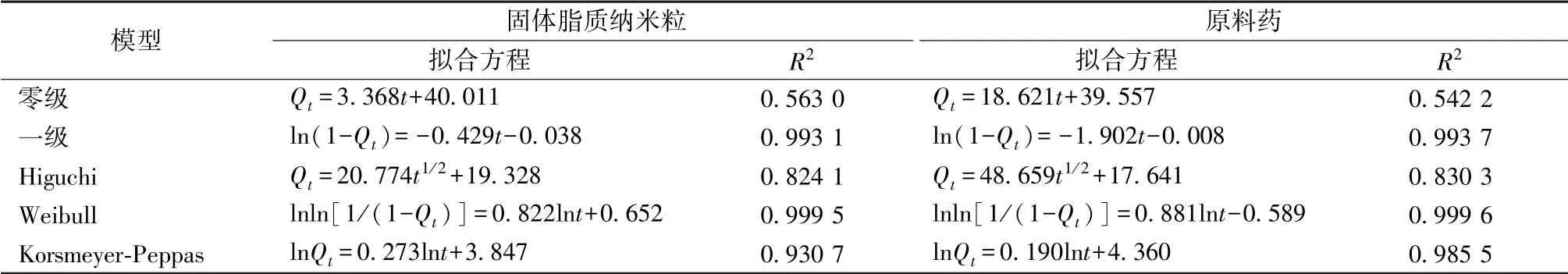
表5 体外释放曲线拟合方程
3 讨论
固体脂质纳米粒的制备方法主要包括高压均质法、 微乳法、 溶剂蒸发法、 乳化蒸发-低温固化法、 薄膜接触器法、 相转换法、 熔融超声法、 高剪切乳化超声法、 溶剂注射法等[13,16]。 根据藤茶总黄酮理化性质及实验室条件, 本实验选择简便易行、 操作可控的熔融超声法, 所得固体脂质纳米粒粒径较小, 均匀稳定, 混悬液具有明显的乳光。
前期对藤茶总黄酮固体脂质纳米粒处方进行考察时,曾选用山嵛酸甘油酯作脂质材料, 其粒径偏大, 均为220 nm左右, 包封率最高达83%; 选用单硬脂酸甘油酯时,粒径变小, 包封率、 载药量均有所提高, 另外以泊洛沙姆188 为乳化剂时药物溶解性好, 安全性高, 故选择单硬脂酸甘油酯和泊洛沙姆作为处方。 与文献报道的藤茶总黄酮主成分二氢杨梅素脂质体[11]、 长循环纳米脂质体[17]相比,本实验所得固体脂质纳米粒包封率(87.27%) 和载药量(12.43%) 均明显提升。
相较于常用的正交设计和均匀设计, 星点设计、 Box-Beheken 响应面法设计精度更高, 可灵敏地考察各因素间的交互作用, 精确找出最佳点, 并且预测性良好。 本实验采用星点设计-响应面法, 通过Design Expert 软件对实验数据进行二项式拟合, 发现模型拟合度较好, 同时验证试验显示该方法预测性良好, 制备工艺简便易行。
体外释放度是评价载药体系的重要指标, 本实验采用经典动态透析法进行考察, 选择零级、 一级、 Higuchi、Weibull、 Korsmeyer-Peppas 等模型进行体外释放数据拟合,发现其释放曲线最符合Weibull 方程。 实验中发现固体脂质纳米粒在初期释放较快, 推测可能是游离药物及固体脂质纳米粒表面吸附的药物共同释放; 后期释放趋于平缓, 可能与药物从载体骨架中扩散, 或随载体材料降解而释放相关。 而且与原料药相比, 固体脂质纳米粒具有一定的缓释特性。
藤茶总黄酮开发前景良好, 为了改善其生物利用度,制备相关制剂是亟待解决的问题之一。 本实验采用熔融超声法制备藤茶总黄酮固体脂质纳米粒, 后续将对其质量进行全面评价, 并进行体内、 体外抗癌药效学、 药代动力学等研究, 以期进一步提高其口服生物利用度及抗癌靶向性。
