银屑病与STAT3
2019-08-25陈怡雯苏婷苏忠兰
陈怡雯 苏婷 苏忠兰
南京医科大学第一附属医院皮肤科 210029
银屑病是一种常见的以红斑、鳞屑为主要临床表现的慢性复发性炎症性皮肤病,病因尚不明确,目前认为与遗传、环境、免疫等多种因素有关。近年发现,银屑病与心脑血管疾病[1]、代谢综合征[2]、精神心理疾病[3]、淋巴瘤[4]及炎症性肠病[5]等具有相关性,因此认为银屑病已不仅仅是皮肤病,更是系统性的炎症性疾病。信号转导与转录激活因子 3(signal transducer and activator of transcription 3,STAT3)是一种重要的转录因子,与肿瘤、自身免疫性疾病等相关。近年研究发现,STAT3在银屑病发病中起关键作用[6],本文就二者的关系作一综述。
一、银屑病发病机制
目前认为,银屑病是在遗传易感基因和环境因素影响下,角质形成细胞(KC)与免疫系统(尤其是T 细胞)共同介导的自身免疫性疾病[7]。银屑病发病具有遗传异质性和种族差异性,遗传因素在其中发挥重要作用,全基因组关联分析(GWAS)迄今累计发现90 个银屑病易感基因[8],它们与抗原提呈、白细胞介素(IL)-23轴、T细胞分化、固有免疫、负性调控等多个银屑病免疫应答环节相关[9]。
环境因素如紫外线暴露、药物、饮食、感染、心理因素等可引起免疫紊乱,在银屑病起病、发展及复发中有重要作用。环境因素可通过与遗传及表观遗传相互作用影响银屑病发病,其中表观遗传学修饰起不可忽视的作用[10]。
近年来一些自身抗原也陆续被发现与银屑病相关。角蛋白17(keratin 17,K17)是唯一可由银屑病相关细胞因子诱导表达的角蛋白,其与链球菌M6 蛋白具有相同的序列ALEEAN,可刺激T 细胞活化和增殖。抗菌肽LL37 不仅能通过IL-23/Th17轴激活树突细胞,还能刺激T细胞介导适应性免疫。它们都在银屑病中高表达,并具有刺激适应性免疫应答的作用,因此被认为是银屑病的自身抗原[11-12]。
T 细胞介导的免疫反应在银屑病发病中具有重要作用。早期研究认为Th1细胞起主要作用,近年来发现IL-23/Th17 轴在发病中占核心地位(图1)。皮肤损伤或感染使KC 释放抗原,激活具有抗原提呈作用的树突细胞,后者迁移至引流淋巴结,产生IL-6、IL-23 介导初始T 细胞向Th17分化,成熟的Th17细胞分泌IL-17A、IL-17F、IL-22等细胞因子,作用于KC 使其过度增殖、异常分化,KC 还能分泌多种细胞因子、趋化因子、抗菌肽,趋化Th17 等炎症细胞至皮肤,从而形成正反馈环路,促进银屑病的发生发展。KC 分泌的血管内皮生长因子(VEGF)具有促进血管生成的作用。近年发现,IL-36通路在脓疱性银屑病发病中具有重要作用,它与IL-23/Th17轴在银屑病发病中相互关联、相互促进[13]。
固有免疫中的中性粒细胞也参与银屑病发病[14],它以细胞外诱捕网的方式分泌细胞因子及炎症介质,代替中性粒细胞发挥长期的免疫功能。
二、STAT3信号转导通路
1.STAT3分子:STAT3存在于细胞质中,可被多种胞外信号激活,进入细胞核影响基因的转录发挥生物学功能,具有信号转导和转录调控双重功能。其有6个功能结构域:N端氨基酸保守序列、螺旋区、DNA结合结构域、连接区、SH2结构域和C 端转录激活区。其中SH2 结构最保守,也是功能最重要的区域。天然存在的STAT3 具有两种形式:STAT3α 和 STAT3β,相对分子质量分别约为 89 000 和80 000,STAT3β在C端缺失一段序列,一般认为它对STAT3通路起负向调控作用。STAT3中两个位点的磷酸化被认为与其转录活性密切相关,一个是705 位酪氨酸位点(Tyr705),另一个是727 位丝氨酸位点(Ser727),后者能使STAT3的转录活性最大化[15]。
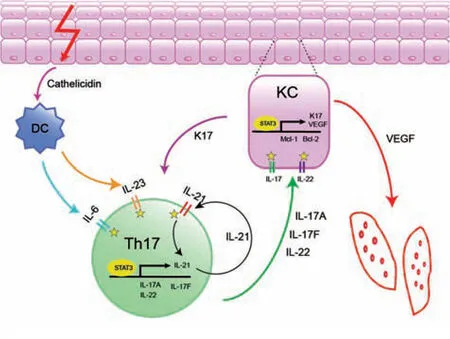
图1 STAT3 在银屑病发病中的作用模式图 皮肤在损伤或感染的刺激下释放抗菌肽,激活树突细胞产生白细胞介素(IL)-6、IL-23等细胞因子,继而激活STAT3,促进IL-21的产生并形成IL-21/STAT3的自分泌环路,促进Th17细胞分化;Th17细胞产生促炎因子IL-17、IL-22等,通过激活角质形成细胞(KC)中STAT3使之过度增殖、异常分化,产生自身抗原K17,进一步激活T细胞形成正反馈通路,分泌血管内皮生长因子(VEGF)促进血管生成。☆示STAT3的激活
2.STAT3 信号通路的激活及调节:STAT3 的激活途径包括:①JAK/STAT途径,是STAT3激活的经典途径,当多种细胞因子或生长因子与靶细胞膜上相应受体结合后,受体发生二聚化并激活受体相关性激酶JAK(主要为JAK1、JAK2和TYK2),受体的酪氨酸残基被JAK磷酸化后进一步招募 STAT3 分子,STAT3 第 705 位酪氨酸进而被 JAK 磷酸化;②丝裂原活化蛋白激酶(mitogen activated protein kinase,MAPK),具有丝氨酸/苏氨酸蛋白激酶活性,可使STAT3 第727 位丝氨酸磷酸化,并使之与Ras 途径相联系,调控STAT3 与靶基因的结合;③非受体型酪氨酸激酶也可激活STAT3[16]。两个磷酸化的STAT3分子通过SH2结构域相互作用形成二聚体,进入细胞核,识别并结合靶基因启动子区GAS 样序列,调控基因转录。STAT3 对基因的调控受细胞类型、转录共激活/抑制因子、募集的转录因子如干扰素调节因子、活化T细胞核因子、核因子κB、叉头状/翼状螺旋转录因子3(forkhead or winged helix transcription factor 3,Foxp3)、SMAD以及细胞因子微环境等因素的影响[17]。
STAT3 的负向调控:①细胞因子信号抑制物(suppressor of cytokine signaling,SOCS)可影响STAT3 的磷酸化,减弱信号传递;②活化的STAT 蛋白抑制剂(protein inhibitors of activated STATs,PIAS)可阻碍STAT3 二聚化及二聚体与DNA 的结合;③蛋白质酪氨酸磷酸酶(protein tyrosine phosphatase,PTP)可使 STAT3 去磷酸化而失活[17];④表观遗传修饰可直接或间接调控STAT3活性,如miRNA、乙酰化修饰等[18];⑤非磷酸化的STAT3(U-STAT3)可结合到DNA序列的AT富集区,调控基因转录[19]。
3.STAT3 的生物学功能:STAT3 参与炎症反应和细胞增殖、存活、凋亡、迁移及血管生成、胚胎发育等多种重要的生物学过程,包括诱导细胞周期调控基因如c-myc、pim-1、cyclin D1 的表达调节细胞增殖,上调抗凋亡基因如Bcl-2、Bcl-xL、Mcl-1等从而延长细胞寿命,抑制细胞毒性T细胞介导的抗肿瘤效应促进肿瘤产生等[20]。正常情况下STAT3的激活迅速而短暂,它的异常活化与肿瘤、自身免疫性疾病如类风湿性关节炎、银屑病等的发生有关[21]。
三、银屑病与STAT3的关系
Sano等[6]发现银屑病患者KC中STAT3异常活化,但其他表现为表皮过度增生的炎症性皮肤病中并没有发现STAT3 表达的异常升高,因此认为STAT3 的活化与银屑病发病相关。他们进一步构建了K5.Stat3C 转基因鼠,其KC可组成性表达STAT3的活化形式——Stat3C,发现小鼠在出生2周内可自发或在外界刺激下发生银屑病样皮疹,其KC中多种银屑病相关基因转录增多,进一步提示STAT3 在银屑病的发病中起重要作用。以下根据STAT3在银屑病不同发病环节中的作用进行分类讨论。
1.参与Th17细胞分化:免疫反应中,初始T细胞的分化方向受病原相关分子模式的种类、抗原数量及T细胞-抗原提呈细胞间的作用强度等因素影响。初始T细胞分化过程中,IL-2和转化生长因子β1分别激活STAT5和SMAD,它们都能诱导Treg 细胞特异性转录因子Foxp3 的表达,此外,SMAD能诱导Th17细胞特异性转录因子维A酸相关孤核受体γt(retinoid-related orphan nuclear receptor γt,RORγt)的表达,至此,初始T细胞发育为Treg 和Th17共同的前体细胞。该前体细胞的最终分化方向取决于微环境中能改变Foxp3和ROR-γt相对丰度的细胞因子,高浓度的转化生长因子β1足以诱导前体细胞分化为Treg,但炎症反应中IL-6 的产生和持续刺激能激活STAT3 通路,进而诱导IL-21 表达,形成IL-21/STAT3 自分泌环路,从而引起STAT3 持续激活,上调ROR-γt丰度,使前体细胞向Th17分化[22]。STAT3在激活状态下,转录因子 RORγt 和 RORα 还可诱导 IL-23R 表达,而IL-23对Th17细胞的存活和稳定有重要作用。Th17细胞分化过程中多种关键细胞因子如IL-6、IL-21、IL-23 的功能都依赖STAT3,而Th17 细胞是银屑病发病中的关键细胞,因此STAT3对银屑病发病有重要作用[22]。
2.参与KC 的过度增殖和异常分化:Th17 和Th22 细胞分泌的IL-22 在银屑病皮肤表型形成中具有重要作用。Zheng 等[23]在小鼠耳部局部注射IL-23,发现表皮增厚及STAT3磷酸化水平升高,敲除IL-22后表皮厚度和STAT3磷酸化水平明显降低,因此推断IL-22 通过激活STAT3 介导IL-23引起的棘层肥厚。Zhang等[24]研究发现,IL-22作用于HaCaT细胞可以激活STAT3,引起细胞增殖,上调抗凋亡相关基因Mcl-1、Bcl-2、存活蛋白及异常角蛋白K16、K17的表达;血红素氧合酶1 通过激活蛋白酪氨酸磷酸酶SHP-1、抑制STAT3磷酸化,从而拮抗IL-22促HaCaT增殖及异常分化的作用。
3.参与KC与炎症细胞的相互作用:K5.Stat3C小鼠能自发或在外界刺激下产生银屑病样皮疹,将其皮肤移植到无胸腺小鼠上,银屑病样损害没有自发或在外界刺激下发生,但局部注射活化的T 细胞后出现了皮疹;将正常小鼠皮肤移植到无胸腺小鼠上,同样局部注射活化的T 细胞却不能诱发银屑病样损害[6]。以上结果提示,STAT3在银屑病皮损活化的KC与T细胞之间起连接作用。
Th17细胞分泌的IL-17A、IL-22可通过激活STAT3诱导KC产生K17[25],K17可进一步作为自身抗原激活T细胞,分泌干扰素γ激活更多KC,在KC与T细胞间形成恶性循环。
IL-36是近年发现的参与银屑病发病的又一促炎因子,由炎症细胞分泌的细胞因子刺激 KC 产生。Müller 等[26]发现,IL-36通过激活STAT3诱导KC中IκBζ的产生,IκBζ的主要功能是调节IL-36下游与炎症反应、中性粒细胞趋化相关的基因,因此形成“炎细胞-KC-炎细胞”作用环路。
4.参与血管形成:血管的异常增生是银屑病的一个重要病理特点。Xia 等[27]研究发现,VEGF 在银屑病皮损血管形成过程中发挥重要作用。VEGF 的持续表达不仅促进皮肤血管生成,还引起表皮及炎症细胞的改变,在银屑病中具有重要作用。另有研究发现,STAT3 的持续激活能上调VEGF 表达,VEGF 启动子区STAT3的结合位点突变后这种作用消失,干扰STAT3信号可以下调VEGF表达,染色质免疫沉淀证实STAT3与VEGF启动子直接结合[28],表明STAT3直接调控VEGF的表达。
综上,在银屑病表皮过度增生和异常分化、真皮血管异常增生、炎细胞浸润等病理改变的形成中,STAT3都发挥十分关键的作用。
四、针对STAT3的治疗
银屑病的治疗以外用药物为主,包括糖皮质激素、维A酸类、维生素D3衍生物、钙调磷酸酶抑制剂等,这些治疗对大多数患者有一定疗效,但容易反复发作,长期外用糖皮质激素还会出现皮肤萎缩、毛细血管扩张、紫癜等不良反应,因此需要寻找更有效且不良反应小的药物。由于STAT3在银屑病发病和维持中有重要作用,因此STAT3 及其上游的JAK激酶可能成为治疗银屑病的有效靶点。
Miyoshi 等[29]利用 STAT3 的小分子抑制剂 STA-21 进行体内实验,发现在K5.Stat3C小鼠皮损处外用含STA-21的软膏,表皮增生几乎完全被抑制,真皮淋巴细胞浸润和毛细血管增生也受到抑制;对8例银屑病患者进行局部治疗,其中6 例在治疗2 周后皮损得到缓解。目前STAT3 抑制剂还在研究中,尚没有用于临床的药物。
miRNA 是一类小的非编码RNA,可以在转录后水平调控基因表达。IL-17 可上调 miR-203 的表达,miR-203 可以抑制SOCS3的表达引起STAT3过度激活,并且上调IL-17引起的 VEGF 分泌,促进银屑病发生[30]。miR-145-5p 可下调STAT3 的表达,对KC 增殖和分泌趋化因子起负向调控作用[31]。通过调节miRNA 从而间接调控STAT3,或许能对银屑病发挥治疗作用[30]。
口服的JAK1、JAK3抑制剂托法替尼[32]和外用的JAK1、JAK2抑制剂卢索替尼[33]已分别完成Ⅲ期和Ⅱ期临床试验,并显示出较好的有效性、安全性及可耐受性。目前观察到的不良反应主要包括鼻咽炎、低密度脂蛋白升高、中度淋巴造血系统抑制等,其长期应用的安全性还有待观察。与以往针对发病机制中某些环节(如T 细胞、肿瘤坏死因子α、IL-23、IL-17等)的生物制剂相比,针对这些关键因子共同汇聚点STAT3的治疗或许可以从多靶点阻断银屑病中复杂的炎症性环路,从而获得更好的治疗效果。
五、结语
银屑病发病机制复杂,目前认为有遗传、环境、免疫的共同参与。本文综述了STAT3 在银屑病发病中的重要作用,为以STAT3为靶点治疗银屑病的可行性提供了依据,但其应用的有效性及安全性尚需进一步研究。
利益冲突所有作者均声明不存在利益冲突
