乌甘胶囊的定性定量研究
2019-08-15孙长河贺康洪
孙长河,贺康洪,赵 昕*
0 引言
原解放军第406医院配制的非标准制剂乌甘胶囊,由海螵蛸[1]、甘草[2]等药味组成,有收敛止血、制酸止痛、解痉敛疮的功效,主要用于胃溃疡、十二指肠球部溃疡及慢性胃炎等症的治疗,临床疗效良好。为保证临床用药有效、安全,本研究对乌甘胶囊的主要成分进行了定性定量方法研究,现报道如下。
1 仪器与试药
日本岛津LC-2010AHT高效液相色谱仪(紫外检测器);KQ3200E型昆山超声波处理器。甘草对照药材(中国食品药品检定研究院,批号:121272-201404),甘草苷对照品(中国食品药品检定研究院,供含量测定用,含量93.7%,批号:111610-201106);样品:乌甘胶囊(规格:0.43 g/粒,原中国人民解放军第406医院配制,批号:140912、140913、140915);试剂:乙腈(色谱纯,Tedia公司),自制纯化水;其他试剂为分析纯。
2 方法与结果
2.1 显微鉴别 显微镜下应可见不规则透明薄片或碎块,具细条纹或网状纹理(海螵蛸)。纤维束周围薄壁细胞含草酸钙方晶,形成晶纤维。具缘纹孔导管较大,稀有网纹导管;木栓细胞红棕色,多角形(甘草)。如图1。
2.2 甘草的薄层色谱鉴别[3]取乌甘胶囊内容物2 g,加稀盐酸3 ml,三氯甲烷15 ml,加热回流1 h,放冷,滤过,蒸干,残渣加乙醇0.5 ml,溶解,作为供试品溶液。取甘草对照药材1 g,加稀盐酸3 ml,三氯甲烷15 ml,加热回流1 h,放冷,滤过,蒸干,残渣加乙醇0.5 ml,溶解,作为对照药材溶液。再按乌甘胶囊处方工艺,除甘草外各味药材制成胶囊,取内容物,按供试品溶液方法操作,制得阴性样品溶液。取上述3种溶液各5 μl,分别点于同一以羧甲基纤维素钠为黏合剂的硅胶G薄层板上,以石油醚(30~60 ℃)-甲苯-乙酸乙酯-冰醋酸 (10∶20∶7∶0.6)为展开剂,展开,取出,晾干,喷以10%磷钼酸乙醇试液,在105 ℃加热至斑点显色清晰,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。阴性溶液无干扰。见图2。
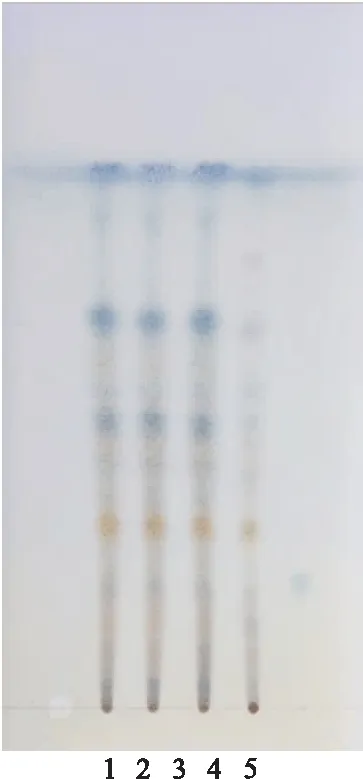
图2 乌甘胶囊TLC鉴别图
2.3 乌甘胶囊中甘草苷含量测定[4-5]
2.3.1 对照品、供试品及阴性样品溶液的制备 ①甘草苷对照品溶液的制备:取甘草苷对照品10 mg,精密称定,至50 ml容量瓶中,加70%乙醇溶解并稀释至刻度,作为对照品储备液。精密量取对照品储备液10 ml,至100 ml容量瓶中,加70%乙醇稀释至刻度,摇匀,即得1 ml含20 μg的对照品溶液。②供试品溶液的制备:精密称取乌苷胶囊内容物0.25 g,置25 ml容量瓶中,精密加入70%乙醇25 ml,密闭,称重。超声处理30 min,放冷,称重,用70%乙醇补足减失的重量,摇匀,取上清液薄膜滤过,即得。③阴性样品溶液的制备:取除甘草外的其他药材,按乌甘胶囊处方比例及制剂工艺制成胶囊剂,按上述供试品溶液制备方法操作,制成阴性样品溶液。
2.3.2 色谱条件与系统适用性 色谱柱:WondaSil C18(4.6 mm×250 mm,5 μm);以乙腈为流动相A,以0.1%磷酸溶液为流动相B,按表1梯度洗脱;检测波长为 276 nm;流速1.0 ml/min;柱温25 ℃。理论板数按甘草苷峰计算不低于2 000。分别取上述3种溶液各10 μl进样,结果表明,样品中待测成分分离良好,阴性对照无干扰,见图3。

表1 甘草苷测定梯度洗脱表
2.3.3 甘草苷对照品标准曲线 精密称取甘草苷对照品适量,加70%乙醇制成每1 ml含20.614 0 μg的溶液,分别精密吸取该对照品溶液2l、6l、10l、14l、18l、22 μl,分别注入液相色谱仪,测定。以进样量为横坐标,以峰面积积分值为纵坐标作图,得一条直线。回归方程为y = 2×106x+52.038,r=1。结果表明,甘草苷进样量在0.041 23~0.453 51 μg范围内,进样量与峰面积呈良好的线性关系。
2.3.4 进样精密度考察 取同一对照品溶液,重复进样6次,计算相对标准偏差,则RSD为0.33% (n=6),符合要求。
2.3.5 中间精密度试验 在同一实验室,两人于不同时间分别用Agilent TC-C18(4.6 mm×250 mm,5 μm)色谱柱与岛津WondaSil C18(4.6 mm×250 mm,5 μm)色谱柱进行试验,按“2.3.1”项下供试品溶液制备方法操作,各制备6份供试品溶液,进行试验。结果表明,中间精密度良好,RSD=1.71%(n=6)。
2.3.6 稳定性试验 取同一供试品溶液(按“2.3.1”项下供试品溶液制备方法操作制备),分别于0、4、8、12、16、24 h进样10 μl,计算相对标准偏差,RSD为0.61%,结果表明,样品溶液至少在24 h内稳定。
2.3.7 重复性试验 取批号为140915乌甘胶囊内容物适量,6份,精密量取,按“2.3.1”项下供试品溶液制备方法操作,制备供试品溶液;另精密称取甘草苷对照品适量,2份,按“2.3.1”项下对照品溶液制备方法操作,制备对照品溶液。测定甘草苷的含量,结果显示,RSD为0.71%(n=6),符合要求。
2.3.8 加样回收率试验 精密称取乌甘胶囊(批号:140915,甘草苷含量为2.152 1 mg/g)0.20、0.25、0.30 g 3个梯度样品,每个梯度3份,分别加入50 ml容量瓶中;称取对照品溶液制备项下的对照品储备液(含量:93.7%,10.56 mg)25 ml,置100 ml容量瓶中,70%乙醇定容至刻度,摇匀,分别取8.0、10.0、12.0 ml对照品溶液加入上述已称取乌甘胶囊的3个梯度容量瓶中,再分别在3个梯度量瓶中加70%乙醇92、90、88 ml,密闭,称重。超声处理30 min,放冷,称重,用70%乙醇补足减失的重量,摇匀,滤过,即得,进样,测定甘草苷的含量,计算回收率,测定结果见表2。
2.3.9 检验3批乌甘胶囊 按本品质量标准“2.3.1”项下供试品溶液制备方法操作,制备供试品溶液,测定3批乌甘胶囊中甘草苷的含量,结果见表3。结果表明,乌甘胶囊各批次甘草苷的含量基本稳定。
3 讨论
3.1 实验过程中,笔者对流动相乙腈-0.1%磷酸溶液(15∶85)、乙腈-0.1%磷酸溶液(28∶72)、70%乙醇-0.1%磷酸溶液(45∶55)进行了考察,结果乙腈-0.1%磷酸溶液(28∶72)为流动相时,甘草苷峰与其他成分峰分离度>1.5,峰形好,对称因子符合规定。取样品溶液进样10 μl,采集数据120 min,在70 min后仍有色谱峰出现,故采取梯度洗脱分析方法。

图3 乌甘胶囊HPLC图
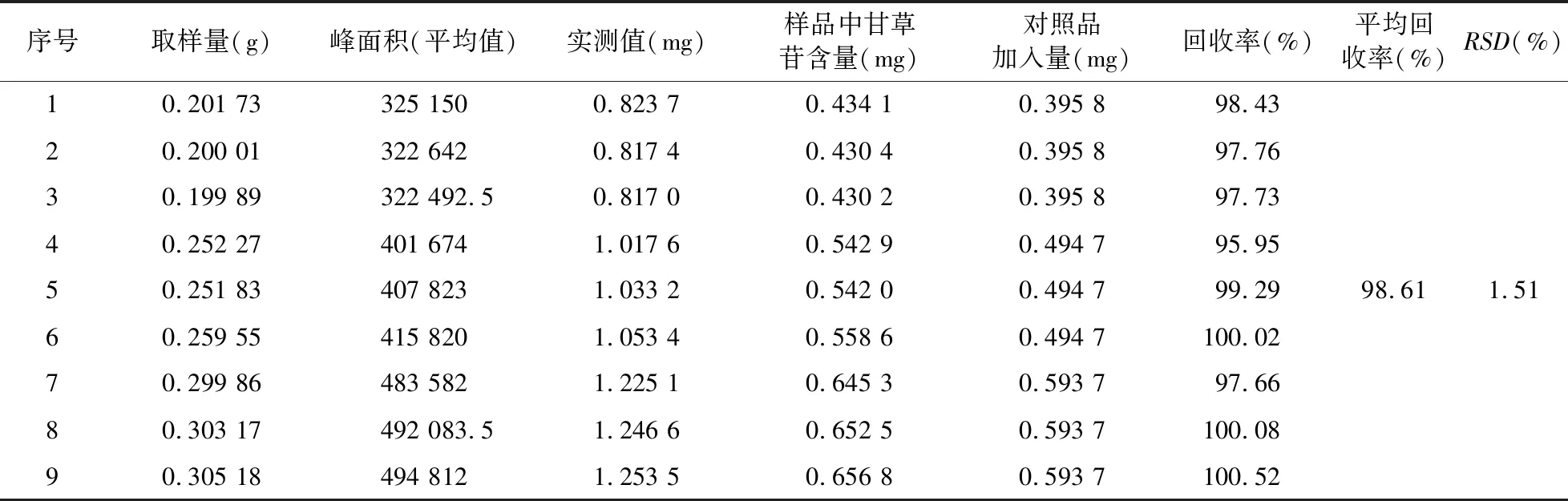
序号取样量(g)峰面积(平均值)实测值(mg)样品中甘草苷含量(mg)对照品加入量(mg)回收率(%)平均回收率(%)RSD(%)10.201 73325 1500.823 70.434 10.395 898.4320.200 01322 6420.817 40.430 40.395 897.7630.199 89322 492.50.817 00.430 20.395 897.7340.252 27401 6741.017 60.542 90.494 795.9550.251 83407 8231.033 20.542 00.494 799.2998.611.5160.259 55415 8201.053 40.558 60.494 7100.0270.299 86483 5821.225 10.645 30.593 797.6680.303 17492 083.51.246 60.652 50.593 7100.0890.305 18494 8121.253 50.656 80.593 7100.52

表3 乌甘胶囊中甘草苷的含量测定结果
注:*为平均值
3.2 制备样品溶液时,为了保证测定准确性,在实验中分别对甘草苷的提取方式、使用的提取溶剂、加入溶剂量、每次提取的时间以及提取的次数进行了考察。其中考察提取方式时,分别采用超声与热回流2种方式,结果表明,采用超声和加热提取方法时,二者所测得的甘草苷含量相当,但超声提取方法的操作简便;考察提取溶剂时,分别使用了50%乙醇、70%乙醇、95%乙醇,结果表明,70%乙醇提取率高,故选取70%乙醇作为提取溶剂。
