新型双功能谷胱甘肽合成酶的真核和原核表达
2019-08-15李佳慧杨芳芳王珍张赛南王首锋
李佳慧 ,杨芳芳 ,王珍 ,张赛南 ,王首锋 ,3*
(1.浙江大学基础医学系,浙江杭州310058;2.绿城农科检测技术有限公司,浙江杭州310052;3.浙江省微生物生化与代谢工程省级重点实验室,浙江杭州310058)
谷胱甘肽是一种由谷氨酸,半胱氨酸和甘氨酸缩合而成的活性三肽,分子式为C10H17O6SN,相对分子质量为307.33,熔点为189~193℃。谷胱甘肽分别以还原性谷胱甘肽(GSH)和氧化性谷胱甘肽(GSSG)形式存在,但在人体内主要为前者。结构决定功能,正是由于其侧链上有一个活泼巯基,能够在生物体内参与许多氧化还原反应,使得谷胱甘肽具有多种生理功能,因此其应用领域也十分广泛。
目前,生产谷胱甘肽的方法有溶剂提取法、化学合成法、发酵法以及酶法。在大多数细胞及革兰氏阴性菌中,谷胱甘肽的合成分2步[1]:第1步,在γ-谷氨酸半胱氨酸合成酶(γ-GCS,大肠杆菌中由基因gshA编码,在酵母中由GSHI编码)的作用下,将L-谷氨酸与L-半胱氨酸合成谷氨酸半胱氨酸;第2步,在γ-谷胱甘肽合成酶(GS,大肠杆菌中由gshB编码,在酵母中由GSHII编码)的作用下,将谷氨酸半胱氨酸合成谷胱甘肽。然而,当体内GSH含量较高时,就会与γ-谷氨酸半胱氨酸合成酶结合,从而降低了GSH的合成与积累。LIAO等[2]通过构建同时含有gshA和gshB基因的重组大肠杆菌,经过表达发现,GSH质量浓度为34.8 mg·L-1,较之前有很大提高。LI等[3]采用2种方法合成谷胱甘肽,一种是在含3种氨基酸和葡萄糖(通过糖酵解提供ATP)的条件下表达酵母细胞,对其生成谷胱甘肽的过程不进行人为干预;另一种是严格控制第1步反应中生成的谷氨酸、半胱氨酸的产量,并加入能恰好消除反馈抑制过程的甘氨酸。结果表明,后者所产谷胱甘肽产量为前者的1.5倍左右。这些均说明谷胱甘肽生成过程中的反馈抑制是可以被降低甚至消除的。
近年来,在一些革兰氏阴性菌和革兰氏阳性菌中发现了一种依赖ATP,能催化谷氨酸、半胱氨酸和甘氨酸生成谷胱甘肽,同时具有谷氨酸、半胱氨酸合成酶与谷胱甘肽合成酶活性的新型酶,即新型双功能谷胱甘肽合成酶GshF。这些细菌主要有Streptococcus agalatiae[4], Streptococcusthermophiles[3], Listeria monocycogenes[5],Pasteurella multocida[6]等。在后2种细菌中,GshF的活性同样受到GSH的反馈抑制。目前,虽已将利用基因改造高产菌株与传统两步法结合进行GSH工业化生产,但仍存在成本较高和反馈抑制等问题,因此,对新型双功能谷胱甘肽合成酶的研究十分重要,其在未来GSH的工业化生产中具有良好的前景。
已有较多实验从Streptococcus thermophiles中克隆目的基因并构建重组表达载体,且GshF表达量较两步法高。刘晓冬[7]从Streptococcus agalatiae中克隆得到目的基因gshF后构建重组表达载体pET-gshFSA,经IPTG诱导表达、超声破胞等处理后测得酶活为0.127 5 U·mg-1。YANG等[8]也从Streptococcus thermophiles中扩增目的基因gshF并构建原核表达载体,然后用IPTG进行诱导表达。迄今,对新型双功能谷胱甘肽合成酶的报道不多,现有报道中多见通过构建原核表达载体对目的基因gshF进行诱导表达。因此,本实验选择从Streptococcus agalatiae中克隆目的基因gshF,再分别将其构建到毕赤酵母和大肠杆菌2种表达载体中,得到重组质粒,分别对这2种重组表达载体进行诱导及酶活测定,最后比较表达产物GshF的酶活大小,选择最适宜且最高效的表达方式,为以后工业生产GSH奠定基础。
1 材料和方法
1.1 材 料
1.1.1 菌株与材料
无乳链球菌(Streptococcus agalatiae)购自苏州北纳创联生物技术有限公司;毕赤酵母GS115,表达载体pPIC9K,质粒pET-28a,大肠杆菌E.coil BL21为本实验室保存。
1.1.2 培养基
LB 培养基(g·L-1):蛋白胨 10,酵母提取物 5,NaCl 10,琼脂粉15(配制固体培养基时加入),使用前加入相应的50 mg·L-1卡那霉素。YPD培养基(g·L-1):酵母提取物 10,蛋白胨 20,葡萄糖 20,琼脂粉20(配置固体培养基时加入)。MD培养基:1.34%酵母基本氮源,4×10-5%生物素,2%葡萄糖,1.5%琼脂。BMGY培养基:1%酵母提取物,2%蛋白胨,100 mmol·L-1磷 酸 钾 (pH 6.0),1.34%YNB,4×10-5%生物素,1%丙三醇。BMMY培养基:1%酵母提取物,2%蛋白胨,100 mmol·L-1磷酸钾(pH 6.0),1.34%YNB,4×10-5%生 物素 ,0.5% 甲 醇 。Todd-Hewitt肉汤(THB)培养基(g·L-1):牛肉粉 10,胰蛋白胨20,葡萄糖2,碳酸氢钠2,氯化钠2,磷酸氢二钠0.4。
1.1.3 工具酶与试剂
高保真DNA聚合酶(PrimeSTAR HS DNA Polymerase)、Taq DNA 聚合酶、SnaB I、Not I、Sal I限制性内切酶均购自宝生物工程(大连)有限公司;质粒小量抽提试剂盒购自Axygen公司;PCR产物纯化试剂盒、T4 DNA连接酶试剂盒、割胶纯化试剂盒购自生工生物工程(上海)股份有限公司;Bradford蛋白定量试剂盒购自BIOMIGA公司。
谷胱甘肽还原型、二硫代双硝基苯甲酸[5,5’-dithiobis-(2-nitrobenzoic acid),DTNB]、丙烯酰胺、甲叉-双丙烯酰胺、G418硫酸盐、考马斯亮蓝R-250、蛋白中分子量marker、50×TAE核酸电泳缓冲液购自生工生物工程(上海)有限公司;测序与引物合成送至生工生物工程(上海)有限公司;试剂和混合溶液的配制方法均参考文献[9]。
1.2方法
1.2.1 试验材料的准备
将购买的冻干标准无乳链球菌(Streptococcus agalatiae)菌种进行复苏,全程均在超净工作台中进行。首先打开冻干菌株瓶并用蘸取75%酒精的棉球擦拭瓶口及瓶身;然后往里加入500 μL标准菌株复溶液,混匀后用接种环挑菌液直接划到THB平板上,37℃培养。
1.2.2 gshF基因克隆
根据TaKaRa公司的MiniBESTBacteriaGenomic DNAExtraction试剂盒,从无乳链球菌(Streptococcus agalatiae)中提取基因组DNA。
根据GenBank公布的新型双功能谷胱甘肽合成酶基因序列(EFV98076)以及毕赤酵母和大肠杆菌表达载体上多克隆位点的特征,分别设计引物对F1/R1和 F2/R2(见表 1)。其中引物 F1含有 SnaB I酶切位点(下划线部分),引物R1含有Not I酶切位点(下划线部分);引物F2含有Not I酶切位点(下划线部分),引物R2含有Sal I酶切位点(下划线部分)。以提取的基因组DNA为模板,分别以F1/R1和F2/R2为引物进行PCR反应以扩增目的基因gshF。PCR 反 应 体 系(50 μL):TaKaRa PrimeSTAR Max Premix(2× )25 μL,正 反 向 引 物 (10 μmol·L-1)各0.5 μL,上述模板 1 μL,H2O 23 μL。
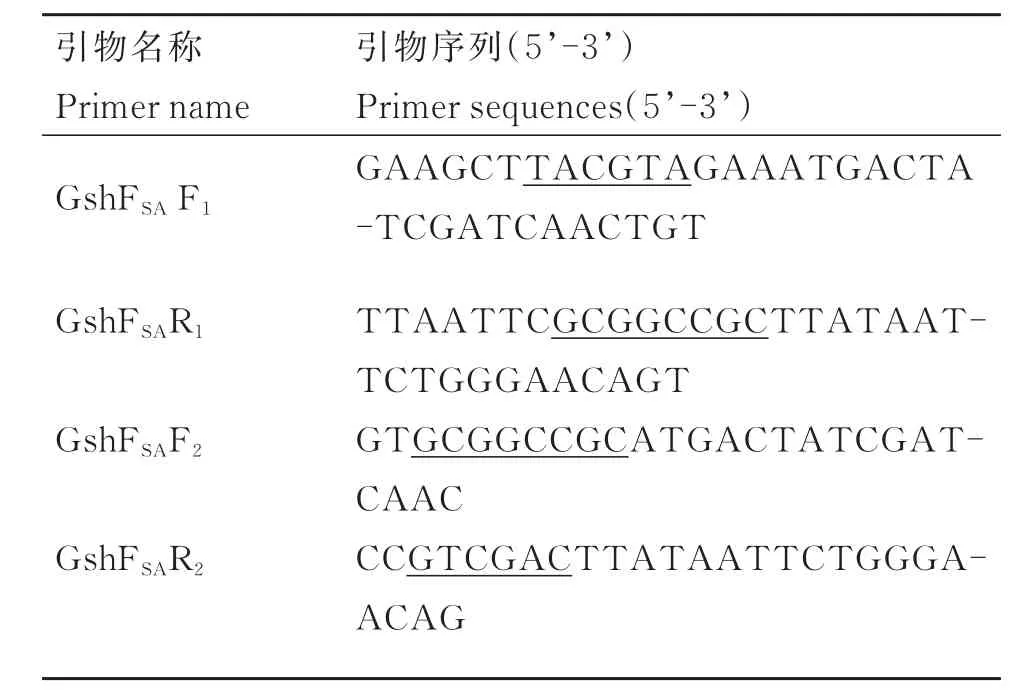
表1 引物Table 1 Primers
PCR扩增程序如下:

随后对PCR产物gshF进行检测与割胶回收。
1.2.3 表达载体pPIC9K-gshF与pET-gshF的构建、转化与检测
本研究采用双酶切的方法将目的基因克隆到表达载体上。由引物对F1、R1扩增得到的目的基因gshF用限制性内切酶SnaB I和Not I双酶切处理后,连接到用相同酶处理的质粒pPIC9K上,得到重组质粒pPIC9K-gshF;将以F2、R2为引物扩增得到的gshF用限制性内切酶Sal I和Not I双酶切后连接到经同样处理的pET-28a上,得到重组质粒pET-gshF。分别热击转化至提前制备好的大肠杆菌感受态细胞BL21后,在含100 μg·mL-1卡那霉素的平板上筛选阳性转化子。
用Sal I限制性内切酶线性化处理构建好的pPIC9K-gshF与空载体pPIC9K,经PCR产物纯化后电转至毕赤酵母GS115中。同时电转空载体pPIC9K作阴性对照。
采用MD平板筛选His+转化子。将重组转化子用高压灭菌水洗,将其分别涂在含0.25,1.0,2.0,4.0 mg·mL-1G418的YPD平板上,进行多拷贝重组菌株筛选[11]。
1.2.4 多拷贝转化子的筛选与鉴定
分别从 0.25,1.0,2.0,4.0 mg·mL-1G418抗性的YPD平板上挑取单克隆于YPD培养基中,震荡培养过夜,取200 μL离心弃上清。再用等体积无菌水洗3次后,取1 μL作为模板进行PCR扩增。经琼脂糖凝胶电泳验证,最终从4.0 mg·mL-1G418抗性YPD平板上鉴定出阳性单克隆。
阳性重组载体pET-gshF可直接诱导表达。
1.2.5 重组酵母GS115/pPIC9K-gshF的诱导表达
将上述阳性重组酵母接种于5 mL YPD液体培养基,30℃过夜震荡培养。按1%接种量转入100 mL BMGY培养基中,30 ℃,250 r·min-1,摇菌培养至OD(600 nm)=2~6。室温下3 000 r·min-1离心5 min后弃上清,用BMMY培养基重悬菌体至OD(600 nm)=1后诱导表达蛋白GshF。期间,每12 h取5 mL培养液且补加无水甲醇至终体积分数为2%。96 h后,收集发酵液,同之前不同时段取出的菌液一起离心,条件为10 000 r·min-1,离心10 min。用三氯乙酸(Trichloroacetic acid,TCA)对上清液脱盐处理后,进行SDS-PAGE检测。
1.2.6 重组大肠杆菌BL21-pET-gshF的诱导培养与破胞
从平板上挑单菌落于含有卡那霉素的5 mL LB液体培养基中,37℃,220 r·min-1培养过夜,然后取1%培养液转接至50 mL含卡那霉素的LB液体培养基中,同条件培养1.5 h后,加入过滤除菌的IPTG至终浓度为1 mmol·L-1,继续同条件培养7 h后,12 000 r·min-1离心后收集菌体[9]。
用 20 mmol·L-1的 Tris-HCl(pH8.0)重 悬 菌体,5 000 r·min-1,离心 10 min,洗涤 3 次。最后将菌体重悬在 10 mL 20 mmol·L-1Tris-HCl(pH8.0),在冰浴中对菌悬液进行超声破胞,破碎条件为:功率200 W,5 s脉冲,7 s停息,共 40次。最后 8 000 r·min-1离心25 min后去掉沉淀,收集粗酶液。
1.2.7 蛋白质量的测定
采用Bradford方法和紫外分光光度法测定[11]。
1.2.8 重组酵母及大肠杆菌表达产物酶活性测定
由于新型双功能谷胱甘肽合成酶可以催化谷氨酸、半胱氨酸和甘氨酸生成谷胱甘肽,谷胱甘肽可由DTNB[5,5-二硫代双(2-硝基苯甲酸)]法测定,且在412 nm具有特征吸收值,因此,可以通过测定吸光值来表征GshF的酶活。酶活测定:采用1 mL反应 体 系 :0.1 mol·L-1Tris-HCl(pH 8.0),40 mmol·L-1Glu,20 mmol·L-1Cys,40 mmol·L-1Gly,100 μL GshF,20mmol·L-1MgCl2,40mmol·L-1ATP,反应液在37℃水浴下反应20 min,取出500 μL与等体积的20%TCA混匀,再在冰上放置30 min后,12 000 r·min-1离 心 10 min,收 集 上 清 测 定 GSH浓度[12-13]。
采用DTNB法测定谷胱甘肽含量。取上述反应所得待测GSH 0.5 mL加入1.5 mL 0.15 mol·L-1的NaOH溶液中,摇匀,再加入3%甲醛溶液(消除Cys的干扰),摇匀并静置2 min。取0.5 mL上述混合液并加入2.5 mL DTNB分析液,摇匀,25℃水浴保温5 min。然后以水为空白对照,测定在波长412 nm处的吸光值[14](DTNB分析液由1体积0.01 mol·L-1DTNB 溶 液 加 100体 积 0.25 mol·L-1(pH 8.0)的Tris-HCl缓冲液配制而成)。从谷胱甘肽的标准曲线上得到对应的GSH浓度,然后计算GshF酶活。
酶活力单位(U)定义为每分钟产生1 nmol谷胱甘肽所需的GshF酶量,用体积比酶活(U·mL-1)来衡量。
2 结果与分析
2.1 生物信息学分析
gshF基因可编码750个氨基酸,蛋白相对分子质量为85 595.51,等电点为5.44。
2.2 gshF基因的克隆
以直接提取的Streptococcus agalatiae基因组DNA为模板,分别利用2对引物和PrimeSTAR Max Premix(2×)扩增目的基因。1%琼脂糖凝胶检测显示,在2 200 bp左右有1目的条带,与预期大小一致(见图1)。将目的片段分别连接在质粒pPIC9K和pET-28a上后送去测序。测序结果显示,扩增得到的gshF与报道的来源于Streptococcus agalatiae ATCC13813的同源性达100%。
2.3 表达载体pPIC9K-gshF和pET-gshF的构建
将目的基因片段gshF分别用相应的限制性内切酶酶切后,连接到用相同限制酶酶切的质粒pPIC9K和pET-28a上,连接产物再热击转化入BL21。重组载体构建完成后再用相应的限制酶双酶切,电泳检测显示,得到约2 200 bp大小的片段(见图2),与预期值相符。

图1 gshF基因的PCR扩增产物Fig.1 PCR product of gshF gene 1:DL5000DNA 标志物;2:gshF基因PCR产物(模板为F1/R1);3:gshF基因PCR产物(模板为F2/R2)1:DL5000DNA marker;2:Amplified PCR product of gshF(F1/R1Primer);3:Amplified PCR product(F2/R2Primer)

图2 重组表达载体pPIC9K-gshF和pET-gshF菌落PCR阳性鉴定Fig.2 The PCR positive identification of pPIC9K-gshF and pET-gshF
2.4 表达载体pPIC9K-gshF的转化与筛选
在将pPIC9K-gshF电转到GS115之前,先用Sal I对其线性化处理。电转至GS115后,用MD平板筛选,结果如图3所示。平板MD-SA为重组载体电转至GS115后所得的重组子,平板MD-pPIC9K为阴性对照。MD-SA上的重组子再分别经含不同浓度G418的YPD平板筛选,最后筛选出对4.0 mg·mL-1G418有抗性的多拷贝重组菌株(见图3)。

图3 重组酵母的G418抗性平板筛选Fig.3 Screening result of G418 resistant plates of recombinant yeast
2.5 重组酵母GS115/pPIC9K-gshF菌落PCR鉴定
用牙签蘸取上述多拷贝单菌落,在10 μL无菌水中震荡后,100℃加热5 min,可直接将其作为模板。用F1、R1(见表1)为引物进行PCR鉴定,如图4所示,在2 200 bp处出现目的条带。
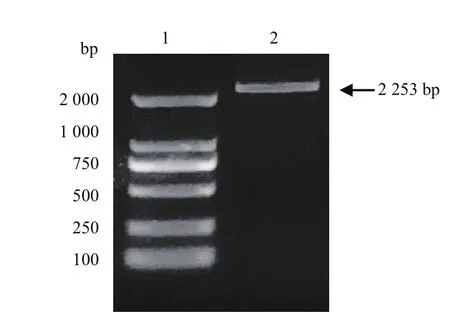
图4 重组gshF抗性酵母菌株菌落PCR阳性鉴定结果Fig.4 Positive identification result of colony PCR of recombinant gshF resistant yeast strain
2.6 重组毕赤酵母甲醇诱导表达
从含4.0 mg·mL-1G418的YPD平板上挑取经PCR鉴定的单菌落,根据Invitrogen公司Pichia expression kit表达说明书,重组菌株以2%甲醇、30℃诱导表达96 h,每12 h取样并添加无水甲醇至终体积分数为2%。离心收集上清液,再经TCA沉淀后,进行SDS-PAGE分析。考马斯亮蓝染色后检测各发酵时间的蛋白表达量(见图5)。从图5可见,在12 h处表达量最高,且在约85 kDa处均有特异性条带,与预期相对分子质量大小相符。

图5 发酵上清蛋白SDS-PAGE分析Fig.5 SDS-PAGE analysis of fermentation supernatant protein
2.7 重组大肠杆菌IPTG诱导表达
从新鲜平板上挑取鉴定过的重组菌株BL21-pET-gshF于含卡那霉素的LB液体培养基中,37℃,220 r·min-1培养过夜,按1%的接种量转接于新的含卡那霉素的LB液体培养基中培养1.5 h,再加入IPTG诱导培养7 h。离心收集菌体,用20 mmol·L-1的 Tris-HCl(pH 8.0)重悬并对其超声处理。经SDS-PAGE电泳与考马斯亮蓝染色后,蛋白表达结果如图6所示。

图6 超声破碎前后蛋白SDS-PAGE分析Fig.6 SDS-PAGE analysis of protein before and after ultra-sonication
2.8 Bradford法测定蛋白含量
按表2所示在不同编号的试管中加入蛋白标准溶液、蒸馏水与Bradford染色液。混匀后静置10 min,测定各管溶液的OD595并以标准蛋白溶液浓度和OD595为横纵坐标绘制标准曲线(见图7),线性回归方程为y=0.089 1x,R2=0.966 2。
取GS115/pPIC9K-gshF发酵表达上清与BL21-pET-gshF诱导表达破壁上清各50 μL加入试管中,再加50 μL蒸馏水。随后加入3 mL Bradford染色液,混匀,室温放置10 min后用紫外分光光度仪测定595 nm处的吸光度。按照标准曲线计算蛋白浓度。其中,GS115/pPIC9K-gshF发酵表达中上清蛋白质量分数为0.46 mg·mL-1,BL21-pET-gshF诱导表达破壁上清中蛋白质量分数为1.46 mg·mL-1。
2.9 谷胱甘肽的检测
精确配制浓度为 0.2,0.4,0.6,0.8,1.0 g·L-1的GSH标准溶液,按照1.2.8节方法测定。以GSH浓度为横坐标,对应的OD412值为纵坐标,绘制标准曲线。所得谷胱甘肽标准曲线如图8所示,线性回归方程为 y=1.081x,R2=0.953 5。
通过测定2种不同表达方式所得GshF酶催化生成的谷胱甘肽在412 nm处的吸光值来计算GshF的酶活,结果见表3。

表2 Bradfrod法测定蛋白所需标准品Table 2 Standards for Bradford
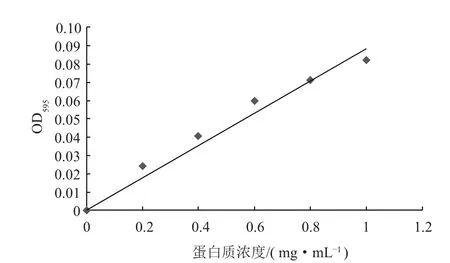
图7 Bradford法测定蛋白标准曲线Fig.7 The standard curve of protein by Bradford

图8 谷胱甘肽的标准曲线Fig.8 The standard curve of GSH

表3 不同菌株发酵表达的GshF酶活分析Table 3 Analysis enzyme activity in different strains
3 讨 论
GSH是一种具有多种生理功能的重要物质,且市场需求量巨大,主要应用在:(1)食品行业。在酒类中添加GSH可维持酒的风味,在面包等中加入GSH可使其保持色泽,加入肉制品中则可维持肉制品的新鲜度等。(2)化妆品行业。利用GSH可清除氧自由基,具有抗氧化等功效,还可用来制备美白产品等。(3)医药行业。可抑制HIV病毒,也可作为抗癌药物等。GSH纯品及其衍生品在全球的销量预计超90亿元[15],因此,降低成本、减少产物抑制显得十分重要。除了在原有两步法生成GSH途径上进行优化外,还可深入研究新型双功能谷胱甘肽合成酶。
近年来,在某些菌中发现了一种同时具备GSH I和GSH II酶活性的新型双功能谷胱甘肽合成酶,且GSH对其不具有反馈抑制作用。与传统两步法生成GSH相比,该方法简化了生产步骤、降低了成本。因此,新型双功能谷胱甘肽合成酶在未来GSH的生产中具有非常好的前景。
常用的表达系统有大肠杆菌与毕赤酵母2种。这2种表达系统各有特点,大肠杆菌表达系统具有生长繁殖快、培养简单、成本低、周期短、表达高的优点,能在较短时间内获得表达产物,但常须对产物进行破胞与纯化,且不具有翻译后修饰功能。毕赤酵母表达系统是最有效的外源蛋白表达体系之一,该系统不但可对表达产物进行加工修饰,使表达的重组蛋白更接近天然蛋白的构象;而且还具有分泌功能,能够将表达产物直接分泌至胞外,减少后期纯化的成本。其高密度发酵生长的特性更适合蛋白的工业化生产。
本实验从无乳链球菌Streptococcus agalatiae中克隆目的基因gshF,利用基因工程的手段构建表达载体pPIC9K-gshF与pET-gshF,然后分别转化到毕赤酵母GS115和大肠杆菌BL21中表达。比较2种不同表达方式所得的GshF酶活,发现GshF更适合在大肠杆菌中表达,且比酶活可达62.15 U·mL-1,而在毕赤酵母表达中的比酶活仅为14.15 U·mL-1。本实验和已有文献的结果表明,gshF可在重组大肠杆菌载体中高效表达,但其在毕赤酵母系统中的表达量较低,可能因无乳链球菌Streptococcus agalatiae为细菌,其来源的目的基因gshF更适合在大肠杆菌系统中表达,但在毕赤酵母表达系统中的密码子具有偏好性。
因此,在后续研究工作中,会对无乳链球菌gshF进行更多分子生物学水平上的功能改造,如基因的密码子优化和利用体外定点突变的方法进行基因的酶活改造,以及后续发酵条件的优化,尤其是对毕赤酵母发酵的温度、pH等条件的优化。同时,还可以利用亲和层析等方法对GshF进行纯化,用来合成GSH和测定其含量。所以,未来在GSH的工业化生产过程中要综合考虑2种表达系统的优劣,通过多角度优化,降低成本,提高产量,以满足市场需求。
