胆木浸膏糖浆的HPLC指纹图谱研究和9种成分的含量测定Δ
2019-08-13刘欢于鑫淼王月王佳月吕星侯丽娟林紫薇孙立新沈阳药科大学药学院辽宁本溪7004本溪市高级中学07级辽宁本溪7000
刘欢,于鑫淼,王月,王佳月,吕星,侯丽娟,林紫薇,孙立新#(.沈阳药科大学药学院,辽宁本溪7004;.本溪市高级中学07级,辽宁本溪 7000)
胆木又名乌檀、熊胆树,为茜草科乌檀属植物乌檀[Nauclea officinalis(Pierre ex Pitard)Merr.et Chun]的干燥枝干、根及树皮[1]。该药味苦、性寒,在《常用中草药手册》《全国中草药汇编》等书籍均有收载,1977年版《中国药典》(一部)也有收录。海南黎族人将胆木作为民间常用草药用于咽喉炎、感冒发热、支气管炎、急性扁桃体炎等疾病的治疗[1-3]。胆木浸膏糖浆由胆木单味药材开发研制而成,具有消肿止痛、清热解毒等功效,临床常用于治疗小儿感冒发热[4]、小儿毛细支气管炎[5]、小儿中耳炎[6]、咽喉炎[7]、急性扁桃体炎[8]等疾病。胆木主要含有生物碱类化合物,也是胆木浸膏糖浆发挥疗效的主要药效组分[9-12]。沈阳药科大学药学院孙立新教授课题组对胆木浸膏糖浆化学成分进行研究发现,除了分离鉴定出短小蛇根草苷、异长春花苷内酰胺和喜果苷等具有解热、镇痛、抗炎活性的生物碱类成分外,还从中鉴定出了原儿茶酸、绿原酸、马钱苷酸等酚酸类化合物和属于环烯醚萜类化合物的獐牙菜苷[13]。酚酸类化合物具有抗氧化、抗炎、抗菌等生物活性,獐牙菜苷具有抑菌、解热作用[13-15],这些酚酸类化合物和环烯醚萜类化合物很可能是胆木浸膏糖浆中除生物碱类成分外发挥临床疗效的重要成分。
目前有关胆木浸膏糖浆的药理作用及其临床应用[4-8]的研究较多,但是对其质量控制研究较少,国家药品标准[16]在含量测定项下要求该制剂总黄酮以无水芦丁计,不得少于2.2 mg/mL,尚不足以全面评价胆木浸膏糖浆的质量。为此,本研究建立了胆木浸膏糖浆的高效液相色谱(HPLC)指纹图谱,并测定其中原儿茶酸、新绿原酸、马钱苷酸、绿原酸、隐绿原酸、獐牙菜苷、短小蛇根草苷、异长春花苷内酰胺和喜果苷共9种成分的含量,以期为胆木浸膏糖浆的质量控制提供参考。
1 材料
1.1 仪器
1260型HPLC仪,包括G7111A 1260 Quaternary Pump VL型四元泵、G7115A 1260 WR型二极管阵列检测器(DAD)、G7129A 1260 Vialsampler型进样器(美国Agilent公司);TG332A型微量分析天平(湘仪天平仪器设备有限公司);KQ5200B型超声波清洗器(昆山市超声仪器有限公司);RE2000A型旋转蒸发仪(上海亚荣生化仪器厂);SHB-Ⅲ型循环水式多用真空泵(郑州长城科工贸有限公司);PB-10标准型pH计(德国Sartorius公司)。
1.2 药品与试剂
胆木浸膏糖浆共20批(批号分别为20161201、20161202 、20161203、20161204、20170101、20170102、20170103、20170506、20170603、20171002、12171102、13171001、12180101、12180102、12180102、13180102、13180103、13171102、13170903、11180307,分别编号为S1、S2、S3、S4、S5、S6、S7、S8、S9、S10、S11、S12、S13、S14、S15、S16、S17、S18、S19、S20,规格:10 mL/支)均购自海南森祺制药有限公司;原儿茶酸对照品(批号:Y-031-161227,纯度:99.07%)、绿原酸对照品(批号:L-007-171216,纯度:99.69%)、马钱苷酸对照品(批号:M-008-180102,纯度:99.36%)、隐绿原酸对照品(批号:Y-067-180425,纯度:99.57%)、新绿原酸对照品(批号:X-014-180410,纯度:99.83%)均购自成都瑞芬思生物科技有限公司;獐牙菜苷对照品(批号:20151208,纯度:98%)购自宝鸡市辰光生物科技有限公司;短小蛇根草苷对照品(批号:20171025,纯度:98.3%)、异长春花苷内酰胺对照品(批号:20180710,纯度:99.9%)、喜果苷对照品(批号:20180615,纯度:98.6%)均由沈阳药科大学药学院药物分析实验室自制;磷酸、乙腈、甲醇均为色谱纯,二氯甲烷、正丁醇均为分析纯,水为纯净水。
2 方法与结果
2.1 色谱条件
色谱柱:Diamonsil C18(2)(250 mm×4.6 mm,5 μm);流动相:乙腈(A)-0.1%磷酸水溶液(B),梯度洗脱(0~10 min,6%A→9%A;10~25 min,9%A;25~30 min,9%A→14%A;30~40 min,14%A→22%A;40~45 min,22%A;45~50 min,22%A→34%A;50~60 min,34%A;60~65 min,34%A→6%A);柱温:30℃;检测波长:240 nm;流速:1.0 mL/min;进样量:10 μL。在上述色谱条件下,取“2.2.1”“2.2.2”“2.2.3”项下溶液进样测定,色谱图见图1。
2.2 溶液的制备
2.2.1 对照品贮备液 取原儿茶酸对照品、新绿原酸对照品、马钱苷酸对照品、绿原酸对照品、隐绿原酸对照品、獐牙菜苷对照品、短小蛇根草苷对照品、异长春花苷内酰胺对照品、喜果苷对照品各适量,置于5 mL量瓶中,用10%甲醇溶解并稀释至刻度,配制成质量浓度分别为 862.0、378.0、770.0、392.0、432.0、398.0、420.0、2 428.0、208.0 μg/mL的混合对照品贮备液,4℃保存,备用。
2.2.2 供试品溶液 精密量取胆木糖浆剂1 mL,置于5mL量瓶中,以水稀释至刻度,摇匀,用0.45 μm微孔滤膜滤过,取续滤液作为供试品溶液。
2.2.3 阴性对照溶液 按样品的制备工艺和配方比例,制备缺胆木药材的阴性样品,再按“2.2.2”项下方法制成缺胆木药材的阴性对照溶液。
2.3 指纹图谱的建立
2.3.1 参考峰的选择 根据图1B可以看出,24号峰在图谱中所占的比例较大,与相邻色谱峰的分离度良好,符合参考峰要求,因此选择其为胆木浸膏糖浆HPLC指纹图谱的参考峰。以24号峰为参考峰,分别计算其他峰相对于24号峰的相对保留时间(RRT)与相对峰面积(RPA),并以RRT与RPA为指标进行方法学考察。
2.3.2 精密度试验 取供试品溶液(编号:S5)1 mL,按“2.1”项下色谱条件重复进样6次,记录各共有峰的RRT和RPA。结果,各共有峰RRT的RSD均小于1.1%(n=6),RPA的RSD均小于2.7%(n=6),表明仪器精密度良好。
2.3.3 稳定性试验 取供试品溶液(编号:S5)1 mL,分别于室温下放置0、2、4、6、8、24 h时按“2.1”项下色谱条件进样分析,记录各共有峰的RRT和RPA。结果,各共有峰RRT的RSD均小于1.2%(n=6),RPA的RSD均小于2.9%(n=6),表明供试品溶液在室温下放置24 h内稳定性良好。
2.3.4 重复性试验 量取胆木浸膏糖浆(编号:S5)各1mL,共6份,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件重复进样分析,记录各共有峰的RRT和RPA。结果,各共有峰RRT的RSD均小于0.76%(n=6),RPA的RSD均小于2.8%(n=6),表明本方法重复性良好。
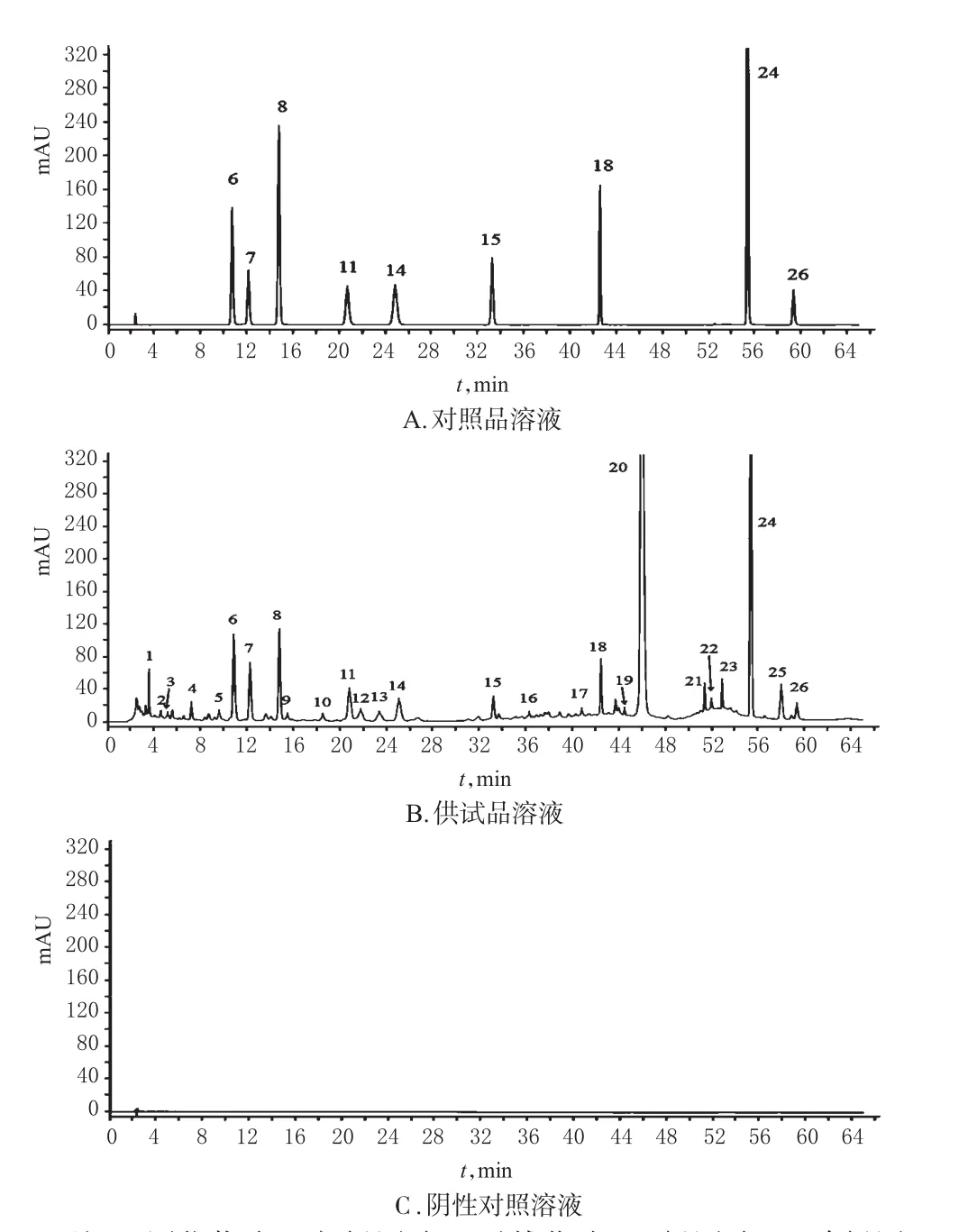
图1 高效液相色谱图Fig1 HPLC chromatograms
2.3.5 不同批次胆木浸膏糖浆的指纹图谱测定与相似度分析 (1)HPLC指纹图谱的生成。精密量取20批胆木浸膏糖浆样品,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样分析,记录色谱图。采用《中药色谱指纹图谱相似度评价系统》(2004A版)对20批胆木浸膏糖浆的HPLC图谱进行分析,得HPLC指纹图谱,详见图2、图3。(2)共有峰的确定与归属。采用《中药色谱指纹图谱相似度评价系统》(2004A版)建立共有模式,绘制对照指纹图谱。通过与对照图谱比对,共标定出26个共有峰。(3)相似度分析。将20批样品指纹图谱与对照指纹图谱进行比较,得到各个指纹图谱与对照指纹图谱的相似度均大于0.98,详见表1。
2.4 含量测定方法的建立
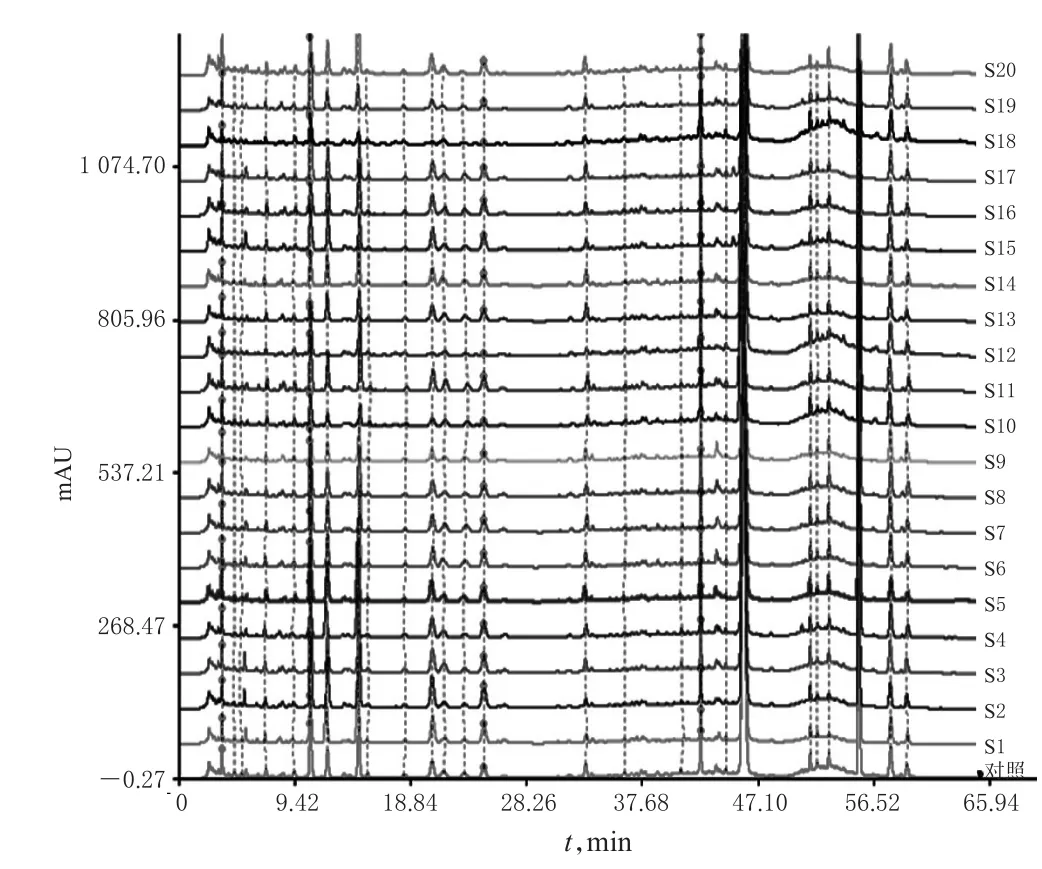
图2 20批样品的HPLC叠加指纹图谱Fig 2 HPLC superimposed fingerprints of 20 batches of samples
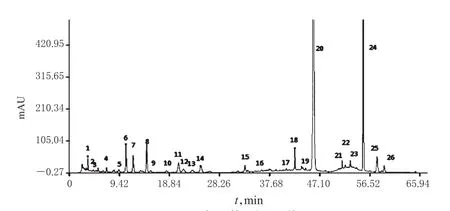
图3 对照指纹图谱Fig 3 Referential fingerprint
2.4.1 系统适用性试验 根据图1所示,供试品色谱峰与对照品各待测物色谱峰的保留时间基本一致,且指标成分在各自对应的位置无干扰,表明该方法专属性良好。所有组分于60 min内检测完毕。在该色谱条件下,各个被测成分之间、被测成分与杂质峰之间得到分离,9种成分的分离度均大于1.5,理论板数以异长春花苷内酰胺峰计大于10 000。
2.4.2 线性关系考察 分别精密量取“2.2.1”项下对照品贮备液 0.1、0.2、0.4、0.8、1.6 mL,置于 5 mL 量瓶中,用10%甲醇稀释至刻度,摇匀,即得系列浓度的混合对照品溶液。分别取上述混合对照品溶液进样分析,记录色谱图。以待测物质量浓度(x,μg/mL)为横坐标、峰面积(y)为纵坐标进行线性回归,得各待测物的回归方程。将对照品贮备液逐级稀释,按“2.1”项下色谱条件进样分析,计算各物质检测限(信噪比为3∶1)及定量限(信噪比为10∶1),结果见表2。
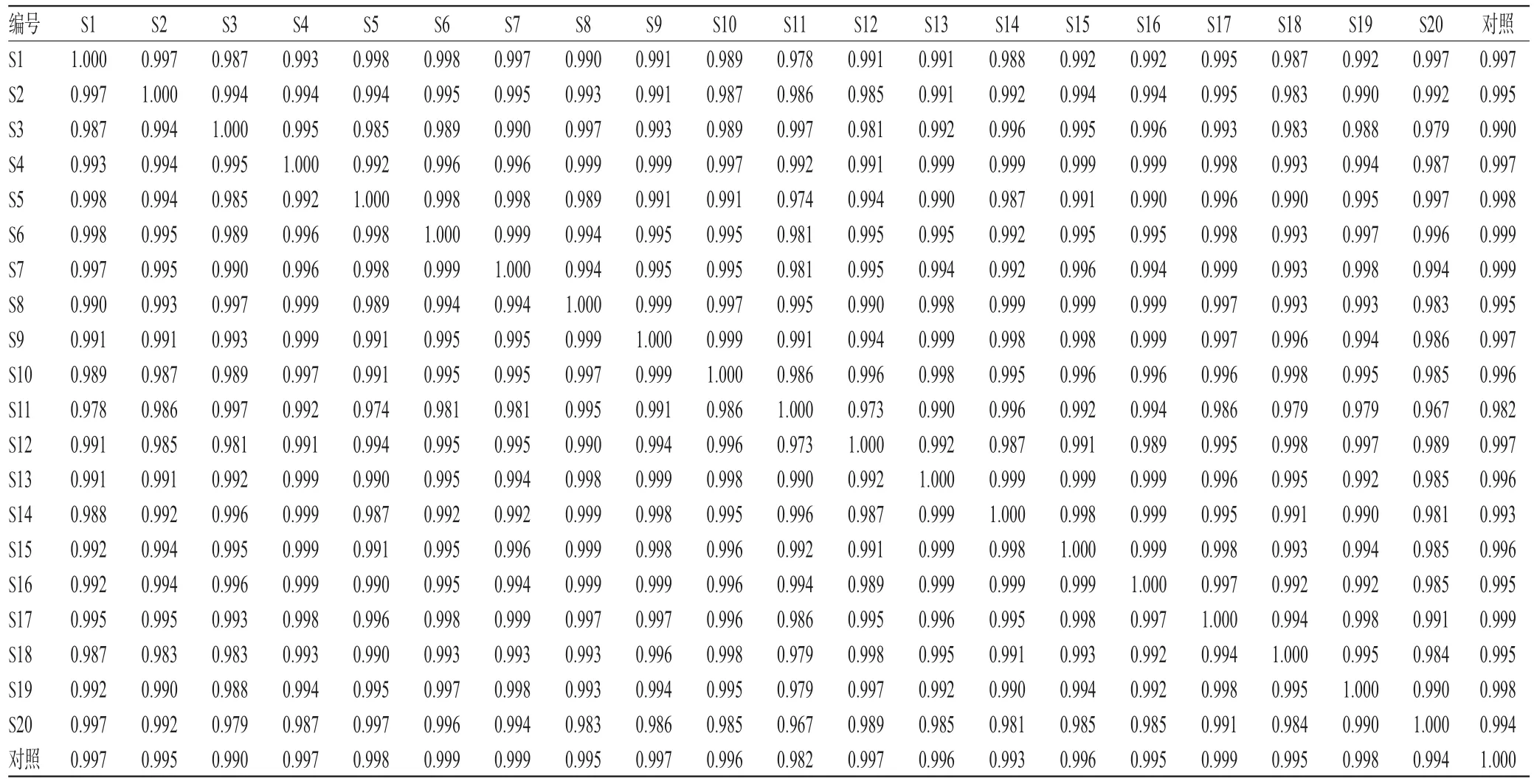
表1 相似度分析结果Tab 1 Results of similarity analysis
2.4.3 精密度试验 取“2.2.1”项下对照品溶液,按“2.1”项下色谱条件重复进样6次,计算原儿茶酸、新绿原酸、马钱苷酸、绿原酸、隐绿原酸、獐牙菜苷、短小蛇根草苷、异长春花苷内酰胺、喜果苷峰面积的RSD分别为0.55%、1.2%、0.34%、0.44%、0.21%、0.23%、0.23%、0.22%、0.67%(n=6),表明仪器精密度良好。
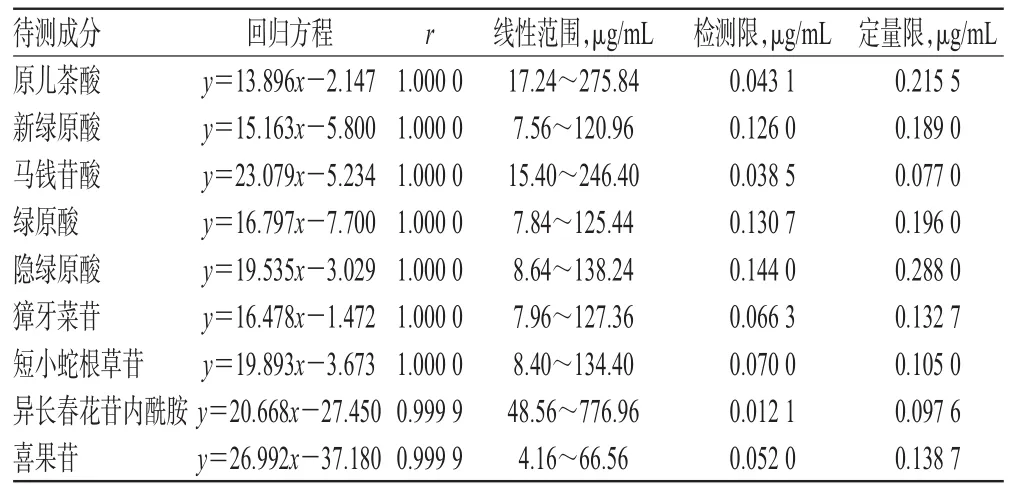
表2 9种待测成分的线性范围及检测限、定量限Tab 2 Linear and ranges,detection limits and quantification limits of 9 compounds
2.4.4 稳定性试验 取供试品溶液(编号:S5),分别于室温下放置0、2、4、6、8、24 h时按“2.1”项下色谱条件进样分析,计算原儿茶酸、新绿原酸、马钱苷酸、绿原酸、隐绿原酸、獐牙菜苷、短小蛇根草苷、异长春花苷内酰胺、喜果苷峰面积RSD分别为0.81%、1.70%、1.20%、0.60%、0.63%、0.85%、1.30%、0.88%、0.99%(n=6),表明供试品溶液在室温下放置24 h内稳定性良好。
2.4.5 重复性试验 精密量取胆木浸膏糖浆(编号:S5)6份,每份1 mL,按“2.2.2”项下方法制备供试品溶液,按“2.1”项下色谱条件进样分析,记录峰面积,按标准曲线法计算含量。结果,原儿茶酸、新绿原酸、马钱苷酸、绿原酸、隐绿原酸、獐牙菜苷、短小蛇根草苷、异长春花苷内酰胺、喜果苷的平均含量分别为479.9、312.8、291.6、258.1、174.1、114.7、125.5、1 768.9、58.4 μg/mL,RSD 分别为0.35%、1.10%、1.70%、0.84%、1.50%、1.50%、1.70%、1.40%、1.30%(n=6),表明本方法重复性良好。
2.4.6 加样回收率试验 精密量取已知含量的胆木浸膏糖浆(编号:S5)6份,每份0.5 mL。分别加入一定质量浓度的对照品溶液(约相当于胆木浸膏糖浆中各成分原有质量的100%),按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进行分析,记录峰面积并计算加样回收率。结果,原儿茶酸、新绿原酸、马钱苷酸、绿原酸、隐绿原酸、獐牙菜苷、短小蛇根草苷、异长春花苷内酰胺、喜果苷的平均加样回收率分别为99.6%、106.3%、100.1% 、102.0% 、98.4% 、100.0% 、99.3% 、100.6% 、101.2%,RSD分别为1.20%、0.24%、0.59%、1.00%、0.73%、1.30%、1.10%、1.80%、1.90%(n=6)。
2.4.7 耐用性试验 (1)色谱柱的考察。精密量取胆木浸膏糖浆(编号:S5)3份,每份1 mL,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件,分别采用Diamonsil C18(2)(250 mm×4.6 mm,5 μm)、Phenomenex C18(250 mm×4.6 mm,5 μm)、Diamonsil C18(1)(200 mm×4.6 mm,5 μm)色谱柱进样分析,记录峰面积,按标准曲线法计算含量。结果,原儿茶酸、新绿原酸、马钱苷酸、绿原酸、隐绿原酸、獐牙菜苷、短小蛇根草苷、异长春花苷内酰胺、喜果苷的平均含量分别为476.8、309.6、291.0、257.0、177.5、115.5、126.1、1 765.5、57.9 μg/mL,RSD 分别为0.91%、1.08%、0.97%、0.98%、1.09%、1.28%、0.92%、0.27%、1.29%(n=3)。(2)流动相中磷酸加入比例的考察。精密量取胆木浸膏糖浆(编号:S5)3份,每份1 mL,按“2.2.2”项下方法制备供试品溶液,分别以乙腈-0.08%磷酸水溶液、乙腈-0.10%磷酸水溶液、乙腈-0.12%磷酸水溶液为流动相,按“2.1”项下梯度洗脱程序进样分析,记录峰面积,按标准曲线法计算含量。结果,原儿茶酸、新绿原酸、马钱苷酸、绿原酸、隐绿原酸、獐牙菜苷、短小蛇根草苷、异长春花苷内酰胺、喜果苷的平均含量分别为 477.1、308.9、290.1、258.0、176.1、115.1、124.5、1 763.4、57.9 μg/mL,RSD分别为0.62%、0.94%、0.76%、0.65%、1.40%、1.18%、1.60%、0.40%、1.68%(n=3)。(3)柱温的考察。精密量取胆木浸膏糖浆(编号:S5)3份,每份1mL,按“2.2.2”项下方法制备供试品溶液,分别将柱温设置为28℃、30℃、32℃,再按“2.1”项下其余色谱条件进样分析,记录峰面积,按标准曲线法计算含量。结果,原儿茶酸、新绿原酸、马钱苷酸、绿原酸、隐绿原酸、獐牙菜苷、短小蛇根草苷、异长春花苷内酰胺、喜果苷的平均含量分别为 476.6、312.6、288.2、257.9、176.8、114.7、125.0、1 766.0、58.3 μg/mL,RSD 分别为 0.74%、0.78%、0.98%、0.70%、0.97%、0.67%、0.76%、0.25%、1.60%(n=3)。
2.4.8 样品含量测定 取20批次胆木浸膏糖浆样品,按“2.2.2”项下平行制备供试品溶液,再按“2.1”项下色谱条件进样分析,记录峰面积并按标准曲线法计算原儿茶酸、新绿原酸、马钱苷酸、绿原酸、隐绿原酸、獐牙菜苷、短小蛇根草苷、异长春花苷内酰胺、喜果苷含量,结果见表3。
3 讨论
3.1 色谱条件的选择
3.1.1 波长的选择 文献[17-19]报道,胆木及其制剂中生物碱类化学成分多在222、227 nm波长处检测。本试验通过DAD优选检测波长,主要考察了在226、240、250nm波长处胆木浸膏糖浆各待测物的色谱行为。结果显示在240 nm波长处,色谱图中色谱峰数较多、峰形较好,故本研究测定波长选择240 nm。
3.1.2 流动相的选择 笔者前期分别对乙腈-水、甲醇-水、乙腈-三乙胺磷酸水溶液、乙腈-甲酸水溶液、乙腈-磷酸水溶液等体系进行了梯度洗脱试验。结果表明,以乙腈-0.1%磷酸水溶液为流动相,所得色谱图基线更平稳,各色谱峰的分离效果最好,故本研究流动相选择乙腈-0.1%磷酸水溶液。
3.1.3 柱温的选择 笔者前期分别对28、30、32、35℃等4种不同的柱温进行了考察。结果表明,30℃时峰形最佳且分离度良好,故本研究柱温选择30℃。
3.2 供试品制备方法的考察
笔者前期分别考察了乙醇加热回流法、乙醇超声提取法、甲醇超声提取法和水饱和正丁醇超声萃取法和水直接溶解法。结果发现,用水溶解,不损失糖浆剂中的各种成分,且专属性良好,因此本研究选择水溶解法作为样品前处理方法。
表3 9种化合物含量测定结果(±s,n=3,μg/mL)Tab 3 Results of content determination of 9 kinds of compounds(±s,n=3,μg/mL)
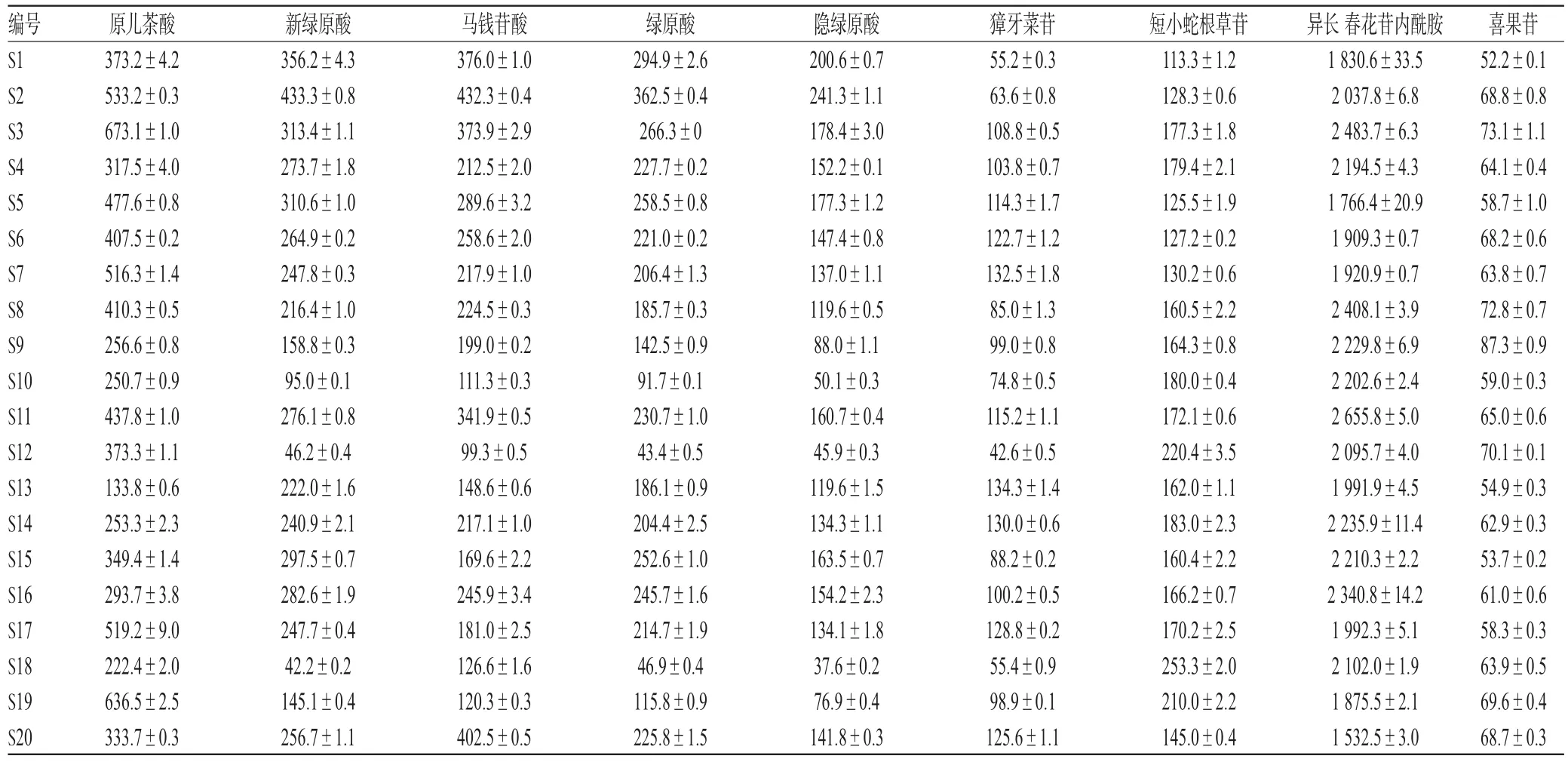
表3 9种化合物含量测定结果(±s,n=3,μg/mL)Tab 3 Results of content determination of 9 kinds of compounds(±s,n=3,μg/mL)
编号S1 S2 S3 S4 S5 S6 S7 S8 S9 S10 S11 S12 S13 S14 S15 S16 S17 S18 S19 S20原儿茶酸373.2±4.2 533.2±0.3 673.1±1.0 317.5±4.0 477.6±0.8 407.5±0.2 516.3±1.4 410.3±0.5 256.6±0.8 250.7±0.9 437.8±1.0 373.3±1.1 133.8±0.6 253.3±2.3 349.4±1.4 293.7±3.8 519.2±9.0 222.4±2.0 636.5±2.5 333.7±0.3新绿原酸356.2±4.3 433.3±0.8 313.4±1.1 273.7±1.8 310.6±1.0 264.9±0.2 247.8±0.3 216.4±1.0 158.8±0.3 95.0±0.1 276.1±0.8 46.2±0.4 222.0±1.6 240.9±2.1 297.5±0.7 282.6±1.9 247.7±0.4 42.2±0.2 145.1±0.4 256.7±1.1马钱苷酸376.0±1.0 432.3±0.4 373.9±2.9 212.5±2.0 289.6±3.2 258.6±2.0 217.9±1.0 224.5±0.3 199.0±0.2 111.3±0.3 341.9±0.5 99.3±0.5 148.6±0.6 217.1±1.0 169.6±2.2 245.9±3.4 181.0±2.5 126.6±1.6 120.3±0.3 402.5±0.5绿原酸294.9±2.6 362.5±0.4 266.3±0 227.7±0.2 258.5±0.8 221.0±0.2 206.4±1.3 185.7±0.3 142.5±0.9 91.7±0.1 230.7±1.0 43.4±0.5 186.1±0.9 204.4±2.5 252.6±1.0 245.7±1.6 214.7±1.9 46.9±0.4 115.8±0.9 225.8±1.5隐绿原酸200.6±0.7 241.3±1.1 178.4±3.0 152.2±0.1 177.3±1.2 147.4±0.8 137.0±1.1 119.6±0.5 88.0±1.1 50.1±0.3 160.7±0.4 45.9±0.3 119.6±1.5 134.3±1.1 163.5±0.7 154.2±2.3 134.1±1.8 37.6±0.2 76.9±0.4 141.8±0.3獐牙菜苷55.2±0.3 63.6±0.8 108.8±0.5 103.8±0.7 114.3±1.7 122.7±1.2 132.5±1.8 85.0±1.3 99.0±0.8 74.8±0.5 115.2±1.1 42.6±0.5 134.3±1.4 130.0±0.6 88.2±0.2 100.2±0.5 128.8±0.2 55.4±0.9 98.9±0.1 125.6±1.1短小蛇根草苷113.3±1.2 128.3±0.6 177.3±1.8 179.4±2.1 125.5±1.9 127.2±0.2 130.2±0.6 160.5±2.2 164.3±0.8 180.0±0.4 172.1±0.6 220.4±3.5 162.0±1.1 183.0±2.3 160.4±2.2 166.2±0.7 170.2±2.5 253.3±2.0 210.0±2.2 145.0±0.4异长春花苷内酰胺1 830.6±33.5 2 037.8±6.8 2 483.7±6.3 2 194.5±4.3 1 766.4±20.9 1 909.3±0.7 1 920.9±0.7 2 408.1±3.9 2 229.8±6.9 2 202.6±2.4 2 655.8±5.0 2 095.7±4.0 1 991.9±4.5 2 235.9±11.4 2 210.3±2.2 2 340.8±14.2 1 992.3±5.1 2 102.0±1.9 1 875.5±2.1 1 532.5±3.0喜果苷52.2±0.1 68.8±0.8 73.1±1.1 64.1±0.4 58.7±1.0 68.2±0.6 63.8±0.7 72.8±0.7 87.3±0.9 59.0±0.3 65.0±0.6 70.1±0.1 54.9±0.3 62.9±0.3 53.7±0.2 61.0±0.6 58.3±0.3 63.9±0.5 69.6±0.4 68.7±0.3
3.3 指纹图谱与含量测定结果分析
本研究在指纹图谱分析中发现26个共有峰,通过与混合对照品比对,共指认出其中9个化合物,分别为原儿茶酸、新绿原酸、马钱苷酸、绿原酸、隐绿原酸、獐牙菜苷、短小蛇根草苷、异长春花苷内酰胺和喜果苷。为考察胆木浸膏糖浆质量,本研究建立了这9种成分含量测定方法,并对20批胆木浸膏糖浆进行定量分析。结果发现,本试验建立的HPLC指纹图谱和含量测定法,方法准确、专属、灵敏。
综上所述,本研究所建立的胆木浸膏糖浆HPLC指纹图谱和定量测定方法分析准确度高、专属性强、灵敏度高,可以为胆木浸膏糖浆的质量控制提供可靠方法。
