浓缩乳蛋白的脱钙处理对高蛋白营养棒质构的影响
2019-07-04余韵刘大松周鹏
余韵,刘大松,周鹏
(江南大学,食品科学与技术国家重点实验室,食品安全与质量控制协同创新中心,江苏 无锡,214122)
高蛋白营养棒(high protein nutrition bar, HPNB)是一种蛋白含量高达15%~50%,水分活度介于0.5~0.8的中间水分食品,其商业配方主要包括蛋白质、糖类及脂肪,一些小分子多羟基化合物如山梨醇、甘油等也会加入其中以控制水分活度,同时起到增塑剂和保湿剂的作用[1]。HPNB应用广泛,但其在储藏过程中发生的理化性质变化极大限制了它的货架期,其中质地劣变最显著。HPNB在储藏后期的质地劣变主要是由蛋白质的自聚集、美拉德反应或相分离所导致,而小分子迁移则是引起储藏前期质地劣变的主要诱因[2-4]。在储藏初期,由于体系中的蛋白相与小分子相间未达到热力学平衡状态,小分子相中的水、增塑剂等将向蛋白相迁移以使体系变得均匀,而这一迁移则会引起体系微观结构的变化,直接表现为体系的质地变化[5-6]。HPNB中的小分子迁移与蛋白配料的水合性质直接相关,而蛋白配料的水合性质又受蛋白分子结构等的影响。
不同种类的蛋白质由于其分子的亲水性不同使得其水合性质存在差别。HPNB的蛋白质来源主要为乳蛋白如乳清分离蛋白(whey protein isolate, WPI)和酪蛋白酸钠(sodium caseinate, NaCN)以及大豆分离蛋白(soyproteinisolate,SPI),其中乳清蛋白的溶解性最好,以其为原料制备的HPNB质地最为均匀柔软[7-8]。浓缩乳蛋白(milk protein concentrate,MPC)作为一种广泛应用于酸奶、冰淇淋、奶酪等乳制品中的新型乳配料,其在高蛋白营养棒中的应用较为少见,主要是由于其溶解性较差,尤其是经储藏后,其溶解性更是显著降低。HOGAN等的研究显示,在以MPC为原料制备HPNB模型体系的过程中,MPC与小分子相混合后,蛋白颗粒无法完全坍塌溶解,使得难以形成均匀体系,其内聚性较差[9]。MPC溶解性较差主要是由于Ca2+会通过离子键介导蛋白和酪蛋白胶束间的非共价聚集,使得MPC表面壳层结构变得致密,从而遏制溶解过程中颗粒的润湿、分散、溶解等步骤,导致其溶解性下降,已有研究表明,通过对MPC进行脱钙处理,可以抑制胶束间的交联,从而提高MPC的溶解性和储藏稳定性[10-11]。然而,将脱钙MPC作为蛋白原料用于HPNB中的研究还较少见,其是否能改善HPNB的质地还有待研究。
本论文通过离子交换制备了一系列不同脱钙率的MPC,以其为蛋白原料制备HPNB模型体系,并将其置于25 ℃下储藏0~14 d,分别通过低场核磁(low-field nuclear magnetic resonance, LF-NMR)、激光共聚焦显微镜、扫描电镜、全质构分析(texture profile analysis, TPA)等方法探究不同脱钙率MPC对HPNB模型体系小分子迁移、微观结构和质构的影响,以拓展浓缩乳蛋白在高蛋白营养棒领域的应用。
1 材料与方法
1.1 实验仪器
超滤系统,三达膜科技(厦门) 有限公司;Spectr AA 220 原子吸收光谱分析仪,美国Varian公司;B-290喷雾干燥器,瑞士BUCHI公司;S3500激光粒度分析仪,美国Microtrac公司;TM-3030扫描电镜,日本Hitachi高科技公司;Heraeus Multifuge XIR冷冻离心机,美国Thermo Fisher Scientific公司;LSM-710激光共聚焦显微镜,德国Carl Zeiss公司;MesoMR23-060V-I低场核磁共振成像仪,苏州纽迈分析仪器股份有限公司; TA-XT-plus质构分析仪,英国Stable Micro System公司。
1.2 实验材料
巴氏杀菌脱脂乳,上海光明集团;罗门哈斯Amberlite SR1L Na树脂,美国Dow’s化学品公司;浓HNO3、浓HCl、HCLO4、氧化镧、叠氮化钠,丙三醇(甘油)、山梨醇,中国国药集团化学试剂有限公司;异硫氰酸荧光素(fluorescein isothiocyanate, FITC),美国Sigma-Aldrich公司。
1.3 实验方法
1.3.1 脱钙MPC的制备及脱钙率测定
以光明优倍脱脂乳为原料,选用10 kDa截留分子质量的聚醚砜超滤膜进行超滤,待脱脂乳浓缩3倍后,进行洗滤(超滤截留液中补加去离子水至原脱脂乳体积后再次超滤),重复洗滤步骤3次,最终将样品浓缩3倍,在获得的截留液中补加0.2 g/L(质量分数)叠氮化钠以抑制微生物生长。参考徐雨婷的脱钙方法及实验结果,根据树脂交换容量和截留液中的钙含量,预设理论脱钙率为0%、10%、30%、45%、60%,在截留液中分别加入0%、1.5%、3%、5%、8%(质量分数)的离子交换树脂,在1 300 r/min转速及室温下离子交换3 h得到不同脱钙率的截留液[12]。调节进出口温度分别为135、75 ℃,气体流量为36 mm, 泵流速为30%对脱钙截留液进行喷雾干燥,得到不同脱钙率的MPC。将所得MPC分装于铝箔袋中,储藏于4 ℃下备用。
取0.15 g备用MPC粉末样品,在其中加入20 mL浓HNO3及5 mL HClO4,高温消化至剩余2~3 mL液体后,用超纯水定容至50 mL。取5 mL定容样品,在其中加入1 mL镧溶液(50 g/L),用超纯水定容至50 mL。稀释后样品用原子吸收光谱分析仪测定其钙含量。
1.3.2 脱钙MPC的微观结构及溶解性表征
采用扫描电镜观察不同脱钙MPC的表面微观结构。取少量样品粉末置于导电胶上,用洗耳球吹去未黏附的粉末后对其进行喷金,在15 kV加速电压下观察并拍照。用激光粒度分析仪测定样品粉末的粒径,在干法模式下测定,颗粒形状设置为“irregular”。
参考HAVEA的方法,称取0.75 g MPC样品,在其中加入15 mL去离子水,在25 ℃下搅拌溶解30 min,取5 mL溶解液直接置于干燥皿中,另取5 mL溶解液在700×g下离心10 min,将离心上清液转移至干燥皿中,105 ℃下烘干18 h,所得上清液固形物含量占溶解液固形物含量的百分比即为样品粉末的溶解度[13]。
脱钙MPC的溶解粒径参考LIU等的方法,采用S3500激光粒度分析仪分别对溶解0和30 min的MPC溶解液进行溶解粒径测定,其中水及蛋白颗粒的折射率分别为1.33和1.57[14]。测试模式为湿法,测量0.02~2 000 μm,颗粒形状为“irregular”。脱钙MPC的溶解液中颗粒的微观结构采用激光共聚焦显微镜进行观察,方法参照MCKENNA的方法并做适当调整[15]。制备2.5 g/L的FITC/乙醇储备液,采用超纯水将其稀释50倍,取0.75 g MPC,用15 mL该稀释液溶解,分别在溶解0和30 min时取样,激发波长488 nm,发射波长510 nm,20倍物镜下观察。
1.3.3 HPNB模型体系的制备
HPNB模型体系的制备参考张靓的方法并做一定调整[16]。模型体系配方(质量分数)为37.0% MPC,28.6%山梨醇,20.0%甘油和14.3%水。将山梨醇、甘油、水先按质量比(10∶7∶5)混合为均匀溶液,在其中加入MPC后混合5 min,使其完全混合均匀,将所得蛋白团揉搓为圆球状,每个圆球1.5 g,直径约为15 mm。将小球置于聚苯乙烯小分装盒中,盖好盖子后用封口膜密封。将小分装盒置于水分活度密闭容器中于25 ℃ 下储藏,根据预实验中HPNB模型体系储藏期间质构变化情况,确定储藏时间为0~14 d,取样时间点分别为0、3、7、14 d。
1.3.4 横向弛豫时间(T2)的测定
采用MesoMR23-060V-I低场核磁共振成像仪测定HPNB模型体系的横向弛豫时间。该仪器的1H振动频率为21.3 MHz。将1.3.3中制备的样品体系用聚四氟乙烯膜包好以防止测试过程中的水分蒸发,样品准确称重,用于结果的标准化。将其置于 25 mm核磁管中,并置于射频线圈中心位置,选用硬脉冲序列进行校准获得中心频率后,选用多脉冲回波序列(CPMG)进行扫描测定,其中回波个数为1 000,重复扫描次数为8次,所有样品在25 ℃下测试,每个样品重复2遍。测试完毕后软件根据“连续分布指数模型”对获得的回波衰减曲线进行反演,得到T2分布图。
1.3.5 微观结构的观察
HPNB模型体系的微观结构用激光共聚焦显微镜进行观察。激光共聚焦显微镜的观察参考JONG等的方法并作调整[17]。配制3.2 mg/mL的FITC/乙醇染色液,将36 μL染色液与1.5 g样品体系混合均匀后,取0.05 g混合物于载玻片上,盖上盖玻片,用AB胶封边防止水分蒸发。将样品载玻片装在铝箔袋中避光。将样品置于25 ℃下储藏0~14 d,分别于0、3、7、14 d取样,激发波长488 nm,发射波长510 nm,40倍物镜下观察。
1.3.6 质构的测定
HPNB模型体系的质构采用全质构分析(TPA)方法测定。选用直径为35 mm的圆柱探头(P35)进行2次下压,下压速度为1 mm/s,触发力5 g,下压比为50%[18]。样品硬度以下压过程中获得的最大应力来表征,破碎力以第1次下压样品破碎时对应的应力来表征,内聚性以第2次下压峰面积占第1次下压峰面积的百分比来表征。同时对样品下压前及下压后的外观形态进行拍照,每个样品重复测定3次。
1.3.7 统计分析
采用SPSS Statistics 20.0软件对数据进行单因素方差分析,检验组间显著性差异,P<0.05表示差异显著。
2 结果与分析
2.1 脱钙MPC的微观结构及溶解性
图1为不同脱钙率MPC的微观结构图,由图1可以看出,不同脱钙率MPC的微观结构没有明显差异,均为表面光滑,略带褶皱的圆球形。

图1 脱钙MPC的微观结构图Fig.1 Microstructures of decalcified MPC
由表1可以发现,颗粒平均粒径大小随脱钙率升高略有减小,但幅度较小,均处于20~22 μm。对于新鲜脱钙MPC,其脱钙率越高,溶解性越好,当脱钙率>28.3%时,其溶解性差异不大,均>98%,这是因为当脱钙率>27%时,MPC中大部分酪蛋白胶束均已完全解离,转变为非胶束态,减少了Ca2+介导的蛋白间的交联,从而使其溶解性提高[8];由图2也可以看出,在相同溶解时间内,随脱钙率升高,蛋白颗粒粒径分布向小粒径方向偏移,溶解液中蛋白颗粒越来越不明显,其中未脱钙MPC在0~30 min均能看到明显完整的蛋白颗粒,脱钙率为18.9%的样品在溶解30 min 后完整蛋白颗粒不明显,脱钙率为28.3%的样品在溶解30 min后基本看不到颗粒,说明颗粒完全溶解;而脱钙率>39.6%的样品则能在一开始便迅速溶解,说明MPC的溶解性随着脱钙率的升高而提高。

表1 脱钙MPC的平均粒径及溶解性Table 1 Mean particle size and solubility of decalcified MPC
注:同一行中小写字母不同代表对应的数据之间差异显著(P<0.05)。
2.2 HPNB模型体系的小分子迁移
LF-NMR可用于检测样品中氢质子在周围化学环境中的分布情况,所得的横向弛豫时间(T2)越小,说明氢质子所受环境束缚越大,自由度越小,也就表明这一部分质子与周围大分子环境结合越紧密,其流动性越弱[19-20]。由于HPNB是一个多相不均匀体系,其中T2分布图常常呈现出多个峰,表明了小分子在体系中的不同分布,储藏过程中T2的变化可以反映出体系中的小分子在不同相间的迁移情况,这一小分子迁移使得体系增塑能力降低,又是导致HPNB在储藏初期微观结构和质地变化的重要原因[21]。图2和图3为以不同脱钙率MPC为原料制备的HPNB模型体系在新制备及25 ℃下储藏0~14 d的T2分布变化图,其中第0天数据为新制备样品在25 ℃下平衡2 h 后测定得到。从中可以发现小分子主要以2种形式存在,其中在0.1~10 ms的主峰T21表示与蛋白质相结合的小分子相中的氢质子,而在10~100 ms的小峰T22则表示处于蛋白相间的游离小分子相中的氢质子[3,8]。未脱钙和脱钙18.9%的模型体系在新制备时其峰分布较宽,尤其是未脱钙样品,表明模型体系不均匀,在平衡2 h后,二者T21峰宽均变窄,说明平衡过程中小分子相和蛋白相间发生融合,使体系慢慢变得均匀;而对于脱钙率>28.3%的样品体系,在新制备好时体系即表现出较为均匀的状态,说明蛋白相与小分子相能快速融合,且随着脱钙率升高,T21主峰越来越窄,说明体系随脱钙率增加而更均匀。在平衡时间内,脱钙率<28.3%的样品体系的T21和T22峰均向左偏移,其值逐渐减小,说明体系中的小分子相一直在向蛋白相迁移;而脱钙率>39.6%的样品体系在制备60 min后,体系即基本达到平衡。对于所有储藏样品体系,其T21值在0~7 d均较稳定,在储藏14 d后有所减小,而T22值则随储藏时间延长而减小,说明在储藏过程中,游离小分子相不断向蛋白相迁移,使得体系流动性降低,且随着脱钙率升高,其值越小,说明脱钙率越高,小分子相与蛋白相结合越紧密(表2)。对于脱钙率28.3%、39.6%、51.0%的样品体系,其T2分布差异较小,这可能是由于这3种脱钙率MPC的溶解性差异不大,使得蛋白相与小分子相结合程度差异较小。随脱钙率升高,体系在储藏过程中小分子分布变化较小,体系更均一稳定,从而有利于保持质构和提高储藏稳定性。
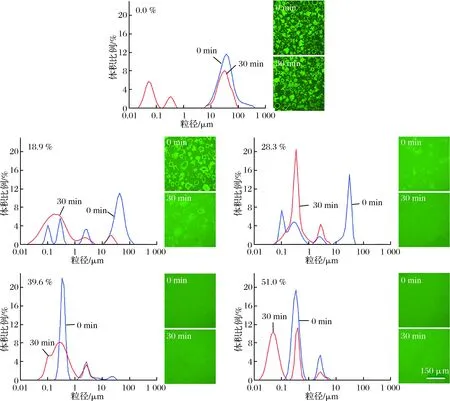
图2 不同脱钙率MPC溶解0和30 min的粒径分布和微观结构图Fig.2 Particle size distributions and microstructures of decalcified MPC at 0 and 30 min of rehydration
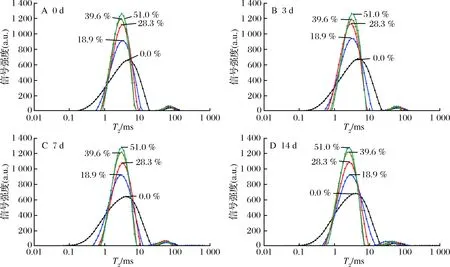
图3 不同脱钙率HPNB模型体系新制备及储藏0~14 d T2分布图Fig.3 Changes in T2 distribution spectra of HPNB model systems formulated with decalcified MPC during 0-14 d storage
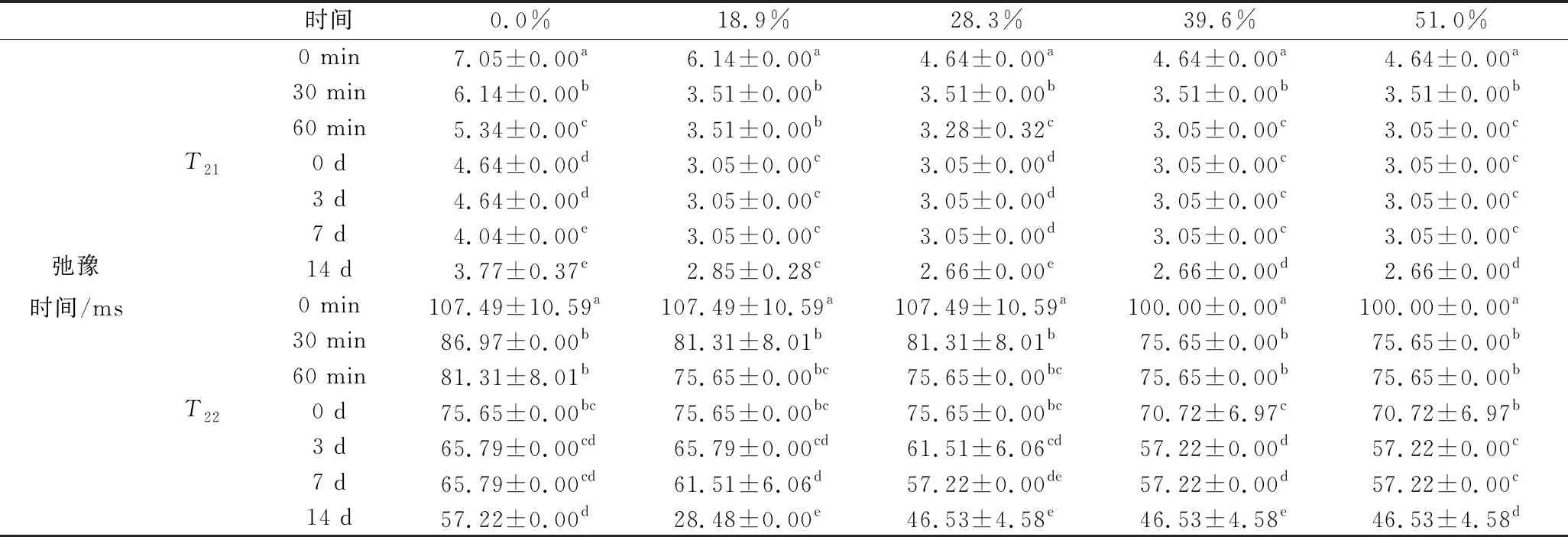
表2 不同脱钙率HPNB模型体系平衡2 h及0~14 d储藏时间内T2弛豫时间常数Table 2 Changes of the T2 peak time of HPNB model systems formulated with decalcified MPC during 2 h equilibration time and 0-14 d storage
注:同一参数同一列中不同小写字母代表对应的数据之间差异显著(P<0.05)。
2.3 HPNB模型体系的微观结构
HPNB模型体系制备过程中蛋白颗粒与小分子相混合的过程也是蛋白颗粒水合的过程,由于不同脱钙率MPC溶解性不同,其体系中小分子迁移速度和程度不同,使得HPNB体系的微观结构存在差异。图4为以不同脱钙率新鲜MPC为原料制备的HPNB模型体系在25 ℃下储藏0~14 d的微观结构变化图。
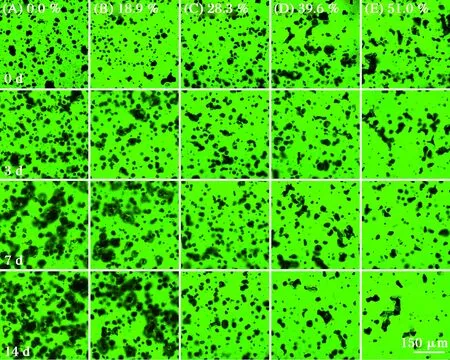
图4 不同脱钙率HPNB模型体系在储藏0~14 d的微观结构图Fig.4 Microstructure changes of HPNB model systems formulated with decalcified MPC during 0-14 d storage
其中,图4中半透明、边缘较暗的不规则区块为部分水合的结构完整的蛋白颗粒聚集体,而绿色荧光较强且均匀的部分为溶解了的蛋白质与水、甘油、山梨醇这些小分子共同组成的连续相[22]。未脱钙样品和脱钙率18.9%的样品体系随着储藏时间延长,蛋白颗粒越来越明显,而这一现象在未脱钙样品中更为显著。在第0天时,脱钙率较低的这2种样品体系中还可以看到典型圆球形带褶皱的蛋白颗粒轮廓,但由于此时溶解了部分蛋白颗粒的小分子相充满了蛋白颗粒的间隙,使得蛋白颗粒形状被掩盖而不明显;随着储藏时间延长,间隙中的小分子相向蛋白相迁移,一部分蛋白溶解变成蛋白碎片,而一些不溶解的蛋白颗粒由于吸收小分子相中的水和甘油而溶胀变大,使得颗粒越来越明显。对于脱钙率为28.3%的样品体系,在储藏时间内,体系较为均匀,仅存在少许蛋白颗粒;而对于脱钙率为39.6%和51.0%的样品体系,在储藏时间内其体系均表现为均匀的连续相,说明其中蛋白颗粒完全与小分子相融合,不存在未溶解的蛋白颗粒。
2.4 HPNB模型体系的质构分析
HPNB在储藏过程中的质地劣变是影响其消费者接受度和货架期的最重要的因素[23]。图5为不同脱钙率HPNB模型体系在储藏0~14 d的TPA结果。
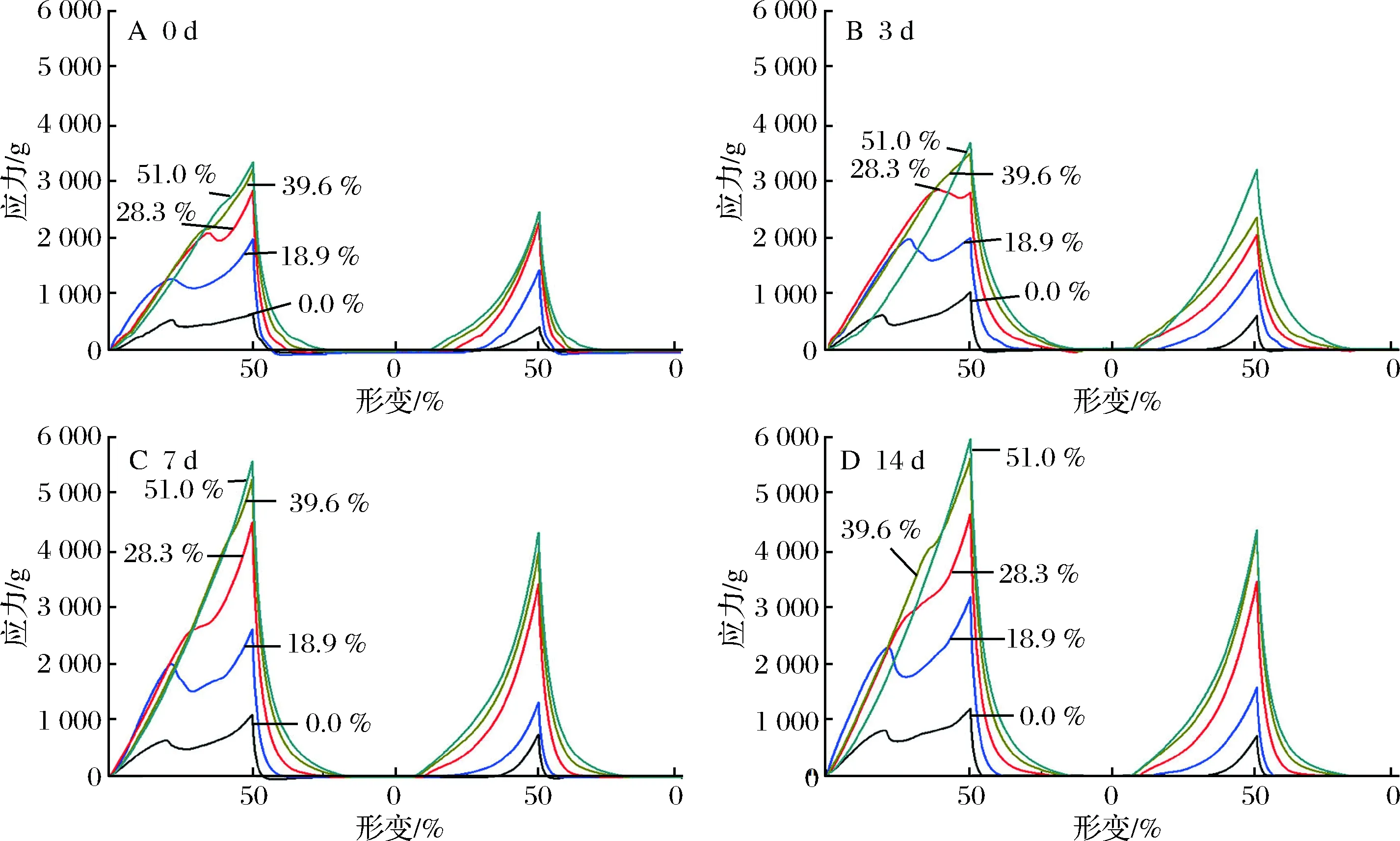
图5 不同脱钙率HPNB模型体系在储藏0~14 d的TPA图Fig.5 Changes in TPA profiles of HPNB model systems formulated with decalcified MPC during 0-14 d storage
由图5可以发现,在相同储藏时间内,脱钙率越高的样品体系硬度越大,所有样品体系硬度均随着储藏时间延长而增加,其中脱钙率为39.6%和51.0%的样品体系硬度始终相接近。对于未脱钙和脱钙率为18.9% 的模型体系,其内聚性随着储藏时间的延长而下降,在实验下压过程中体系易发生碎裂;而对于脱钙率>28.3%的模型体系,其内聚性均随储藏时间延长而有所增加,其中脱钙率为28.3%的模型体系在储藏0~3 d时,体系在下压过程中发生碎裂,而在储藏7 d后,体系在下压时不发生碎裂,而脱钙率>39.6%的样品,体系在0~14 d内聚性均较好,在下压过程中始终能保持结构完整而不发生碎裂(表3、图6)。
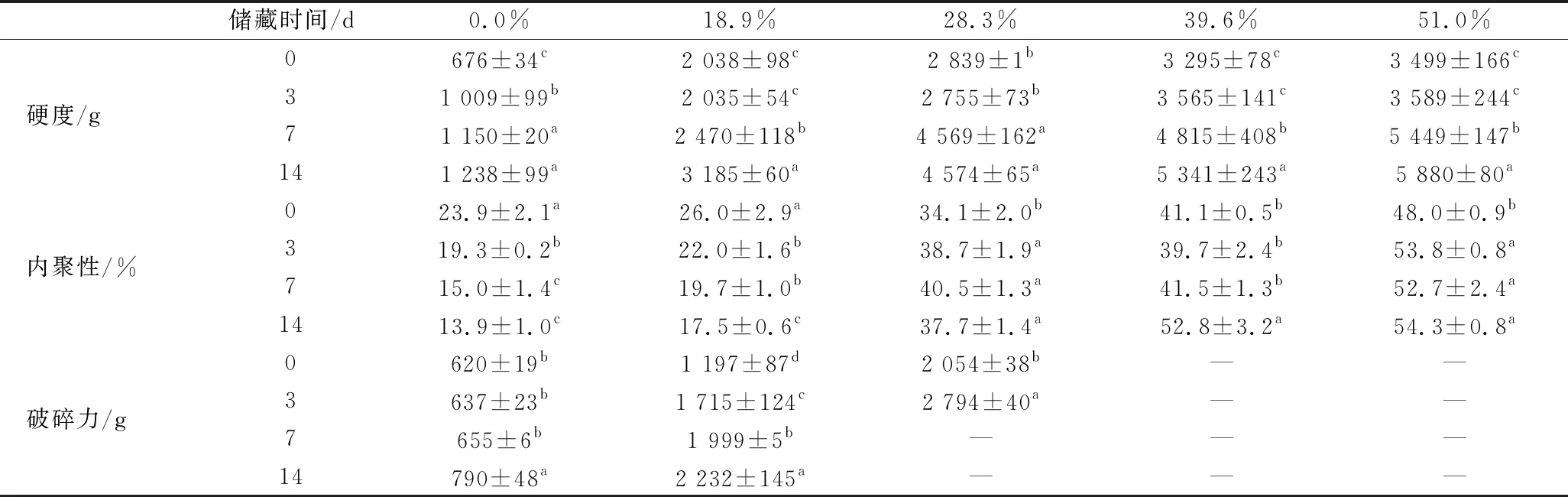
表3 不同脱钙率HPNB模型体系在储藏0~14 d硬度、内聚性及破碎力的变化Table 3 Hardness, cohesiveness and fracturability changes of HPNB model systems formulated with decalcified MPC during 0-14 d storage
注:同一参数同一列中小写字母不同代表对应的数据之间差异显著(P<0.05);“—”代表样品不发生破碎。
如2.2中结果所述,由于在储藏期间小分子向蛋白相迁移,使得体系流动性和增塑性降低,从而导致硬度增大。对于脱钙率<18.9%的模型体系,由于其中存在较多的未溶解的蛋白颗粒,随着蛋白颗粒与小分子相融合,蛋白颗粒溶胀变大,蛋白聚集体间堆积更为紧密,这可能也是导致体系变硬的原因之一。随着脱钙率升高,MPC中的酪蛋白胶束解离程度越大,使得胶束内部疏水基团暴露更多,随着储藏时间延长,蛋白间发生相互作用交联的可能性更大,使得体系硬度更大[24]。HOGAN等选用酪蛋白酸钠、酪蛋白

图6 不同脱钙率HPNB模型体系在储藏0 d和14 d时下压前后外观图Fig.6 Visual appearance before and after compression of HPNB model systems formulated with decalcified MPC at 0 and 14 d storage
胶束、浓缩乳清蛋白、水解乳清蛋白等蛋白原料制备蛋白棒,发现酪蛋白酸钠体系硬度最大而酪蛋白胶束体系则最柔软,即当酪蛋白以非胶束形式存在时,以其为原料制备的蛋白棒硬度较大[7]。在未脱钙和脱钙率为18.9%模型体系中,由于小分子迁移,使得蛋白颗粒间存在的小分子相减少,空隙变多变大,从而使得模型体系更为松散易碎,内聚性较差;而脱钙>28.3%的模型体系由于其在储藏过程中始终为连续均一相,且在储藏过程中蛋白间进一步发生作用交联,从而使得体系呈现出很好的内聚性。当脱钙率<18.9%时,所得模型体系不连续均一且在储藏过程中不稳定,结构松散易碎,不利于产品的储藏和运输;当脱钙率>39.6%时,样品体系内聚性有极大的改善,但硬度较高且钙含量较低,不易被消费者接受;脱钙率为28.3%的样品体系在储藏过程中始终保持均一稳定,且其硬度适中,内聚性好并保留有较多的钙成分,可同时满足运输、储藏及消费者的需求[25]。
3 结论
以脱钙率分别为0.0%、18.9%、28.3%、39.6%、51.0%的MPC为蛋白原料,与山梨醇、甘油、水混合制备HPNB模型体系,探究不同脱钙率MPC对高蛋白营养棒体系小分子迁移、微观结构及质构的影响。MPC溶解性随着脱钙率增加而提高,当脱钙率>于28.3%时,其溶解性均高于98%。在蛋白棒模型体系中,脱钙率越高,小分子迁移的速度越快,小分子与蛋白相间结合越紧密,体系越容易达到均一稳定状态;对于脱钙率>28.3%的模型体系,在储藏7 d后体系间小分子的状态分布差异不大。对于未脱钙和脱钙率为18.9%的模型体系,体系中未溶解的蛋白颗粒随储藏时间延长而越来越明显,而脱钙率为28.3%的模型体系只看到少量的蛋白颗粒,脱钙率>39.6%的体系则在储藏过程中始终为均一的连续相。对于所有HPNB模型体系,其硬度均随储藏时间延长而增加,脱钙率越高,体系硬度越大,但内聚性越好,体系在下压过程中越不容易碎而保持完整的结构。综上所述,以脱钙率为28.3%的MPC为蛋白原料制备HPNB模型体系,其体系均一稳定,硬度能满足储藏要求,内聚性得到极大改善,同时也可较多地保留MPC中的钙成分。研究为拓展浓缩乳蛋白在高蛋白营养棒领域的应用提供了理论基础。
