扩增子测序分析助力传统发酵食品微生物群落研究
2019-06-11孙善峰黄晓宁韩北忠陈晶瑜
孙善峰,黄晓宁,韩北忠,陈晶瑜*
(1.中国农业大学 食品科学与营养工程学院食品质量与安全北京实验室,北京 100083;2.中国农业大学 农业部葡萄酒加工重点实验室,北京 100083)
以分子生物学为基础的生物信息学的诞生和发展最早可以追溯到20世纪60年代,两届诺贝尔奖得主鲍林的分子进化理论的出现,预示着生物信息学的来临[1]。2001年,随着人类基因组测序工作的完成和人类基因组草图的公布,生物信息学的研究走向了一个高潮[2]。生物信息学的根本目标是增加对生物学过程的认识,具体而言,就是发展和应用有效的计算方法(如模式识别、数据挖掘、机器学习算法和可视化技术等)来达到对生物学更深层次的理解[3]。
宏基因组学是专门针对复杂微生物群落发展起来的基因组学技术,其研究基本上可以分为核糖体核糖核酸(ribosomal robonucleic acid,rRNA)基因(细菌和古菌的16S rRNA基因或真菌的18S rRNA、28S rRNA基因和ITS基因)的分类和鉴定(扩增子测序)、功能基因的多样性和分类分析以及全部宏基因组脱氧核糖核酸(deoxyribonucleic acid,DNA)的整体测序(全基因组测序)和分析等[4]。扩增子测序利用独立培养的分子生物学技术、方法和手段对宏基因组进行系统研究,分析微生物在研究对象中的基因序列集合,研究其群落结构与预测功能等。1986年,OLSENGJ等[5]提出,直接从环境中克隆核糖体小亚基RNA编码基因,即16S rRNA基因,开启了以独立培养的分子生物学方法研究微生物多样性的新大门。1996年,STEIN J L等[6]通过构建海水微生物克隆文库,鉴定出了未培养过的古菌的16SrRNA基因,成为宏基因组在微生物领域研究的里程碑。基于宏基因组数据的生物信息学方法广泛用于微生物学研究,根据微生物来源,可以分为土壤微生物、海洋微生物、肠道微生物、农业微生物、食品微生物等。与其他领域相比,扩增子测序数据的生物信息学分析在传统发酵食品微生物研究中的应用起步晚、发展慢,仍存在差距,但最近的相关研究逐渐增多,本文归纳和比较了近几年发展的主要的生物信息学分析方法,以期为今后的研究提供参考。
1 扩增子测序技术在传统发酵食品微生物研究中的优势
中国传统发酵食品风味独特、营养丰富,参与发酵过程的微生物种类众多,形成的菌群结构复杂[7]。面对中国传统发酵食品中如此复杂的微生物,仅凭借低通量、纯培养的方法效率很低。随着扩增子测序技术在微生物研究中的应用以及生物信息学分析的渗入,为复杂微生物菌群多样性的阐明、功能和相互作用的预测提供了更为迅速的解决方案。扩增子测序技术在微生物研究中的应用极大的促进了科研人员对于复杂微生物菌群的认识,其规避了无法培养的微生物对群落中微生物种类认识的局限。自扩增子测序技术应用于传统发酵食品微生物研究以来,产生了大量的生物序列数据,为了充分利用这些序列数据,揭示数据背后隐藏的生物学信息,需要使用信息科学的方法和技术进行管理和分析。生物信息学以低成本和高通量的方式从大量的生物学数据中挖掘出对研究对象有价值的信息,为实验研究提供参考和指导。
2 扩增子测序数据的生物信息学分析
2.1 生物信息学研究概况
生物信息学研究主要包括数据库、算法和工具。核酸数据库按照构建方式有一级和二级之分。最权威的三大国际核酸一级数据库为GenBank[8]、EMBL[9]、DDBJ[10]。二级数据库是基于一级数据库,增加相关信息并经加工和构建而成的具有特殊生物学意义和专门用途的数据库,如能够对三域微生物(细菌、古菌、真核)rRNA基因序列分类的数据库SILVA等。自1994年以来,每年“Nucleic Acids Research”期刊的第一期刊物为分子生物学数据库特刊,回顾当前的分子生物学数据库资源,截至2018年已收录数据库1 898个[2,11]。
生物信息学研究的常用算法有模式分类、统计算法、特征提取、数据压缩等。这些算法在宏基因组数据处理中均有应用,如模式分类中的聚类方法用于挖掘基因之间的调控关系,K近邻法(k-nearest neighbor,k-NN)用于系统发生树的构建;统计算法中的隐马尔可夫模型用于基因预测;特征提取算法用于基于序列特征的宏基因组数据的分装;数据压缩用于主成分分析(principalcomponentanalysis,PCA)等,为实验人员提供了大规模数据分析的手段和实验设计的重要参考。
生物信息学为生物学研究提供了有力的分析工具,通过分析和处理实验数据进行提示、指导甚至替代部分实验操作。基于扩增子测序的宏基因组分析常用生物信息学工具见表1[1]。

表1 基于扩增子测序的宏基因组分析常用生物信息学工具Table 1 Bioinformatics tools commonly used for metagenome analysis based on amplicon sequencing
2.2 扩增子测序数据的生物信息学分析方法
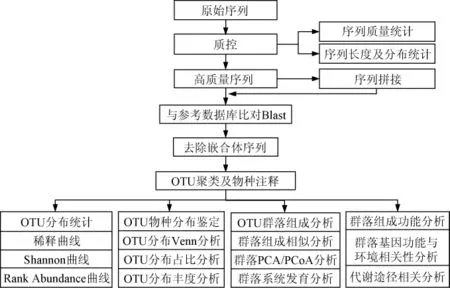
图1 基于扩增子测序的宏基因组数据生物信息学分析流程Fig.1 Bioinformatics analysis process of metagenomic data based on amplicon sequencing
基于扩增子测序的宏基因组数据生物信息学分析基本流程见图1[1],主要包括:提取样本总DNA;聚合酶链式反应(polymerase chain reaction,PCR)扩增目标区域;构建质粒文库测序;测序数据去噪处理(如去除接头、序列标签、引物序列、低质量的序列及嵌合序列等);去噪序列聚类生成分类单元(operational taxonomic units,OTUs),并进行后续生物信息学分析(OTU代表序列分类学注释、系统发育树绘制及多样性分析等),同时可以结合实时荧光定量聚合酶链式反应(real-time quantitative polymerase chain reaction,RT-qPCR)进而实现菌群分布定量及差异比较分析。目前,主流的菌群鉴定软件QIIME和MOTHUR都是按照图1流程实现的[21-22]。QIIME无论在时间消耗,还是在聚类结果的准确性及可信性上,都优于MOTHUR聚类方法,更适合高通量测序数据和复杂环境样本[1]。2018年,QIIME2.0正式上线,QIIME2.0中DADA2[15]代替了原先的UCLUST。DADA2根据扩增子测序数据推断确切的扩增子序列变体(amplicon sequence variant,ASV),取代较粗糙的操作分类单元(OTU)聚类方法;DADA2改善了菌群多样性和差异性的下游分析,并可能使扩增子测序方法探测到菌株水平的变化;DADA2通过准确重建扩增子测序的群落提高了研究人员对微生物群落的研究,比较表明,DADA2比MOTHUR[21]、QIIME(UCLUST)[22]、UPARSE[23]、MED[24]方法更准确[15]。今后,QIIME2流程将会越发广泛的应用于扩增子测序结果分析。
通过聚类获得的OTU代表序列,需要与相应数据库比对注释以进行下一步的分析。目前微生物学研究常用的数据库有rRNA基因数据库(RDP、SILVA、Greengenes、EzBioCloud等)和内部转录间隔(internal transcript space,ITS)序列数据库(UNITE、ITS2等)。RDP和SILVA是目前微生物学研究rRNA基因比对注释常用的数据库。RDP的Classifier功能可用于rRNA基因测序后的物种分类鉴定,能够方便地确定某条rRNA基因序列从门到属/种水平的分类信息并给出各水平相应的置信度。SILVA结合ARB软件进行rRNA质量检测和序列比对,是MOTHUR分析工具中推荐的数据库。Greengenes和EzBioCloud是仅收录16S rRNA的数据库。Greengenes数据库收录有微生物的功能信息,结合PICRUST分析工具能够对微生物群落功能进行预测。对于EzBioCloud,主要是利用数据库的Identify功能确定16S rRNA基因序列的近缘可培养或模式种,较少用于高通量测序16S rRNA的参比。UNITE(仅收录ITS序列)和ITS2(仅收录ITS2序列)是真菌的专属数据库。UNITE和ITS2在应用上有所区别,前者通常用于分类注释,后者通常用于真菌鉴定。
OTU聚类及物种注释后的分析由于研究目的不同会呈现出一定的差异。但核心分析主要包括两个方面:物种组成分析和功能组成预测分析。群落的组成分析、系统发育分析、相似性分析、PCA/主坐标分析(principal co-ordinates analysis,PCoA)是常用的物种组成分析方法。功能组成预测分析主要包括群落组成功能预测分析、群落基因功能与环境相关性分析、代谢途径相关分析等。一般分析思路为:对物种或功能先进行整体描述,然后分析总体差异和分组差异,最后进行微生物组和环境因子关系的关联分析并用无菌体系进行验证以证实因果。
Microbiome Helper[47]是一款整理优化了多套扩增子(16S/18S/ITS)、metagenome分析流程的开源软件。作者提供了一系列的辅助脚本,使宏基因组数据分析更加自动化,显著提高使用者的分析效率。2018年6月发表的基于Galaxy的微生物数据分析框架ASaiM[26]集成了100多种分析工具,内置了多种参考分析流程,能够通过Docker方式快速部署,方便非组学分析研究领域的使用。
3 扩增子测序数据生物信息学分析在传统发酵食品微生物研究中的应用
3.1 生物信息学分析方法在传统发酵食品微生物研究中的常见应用
现阶段,采用基于扩增子测序的宏基因组学方法研究中国传统发酵食品微生物,旨在识别、鉴定传统发酵食品微生物的菌群结构,预测代谢功能,阐明相互作用关系。基于扩增子测序的宏基因组数据生物信息学分析方法在传统发酵食品微生物研究中的代表性成果见表2。
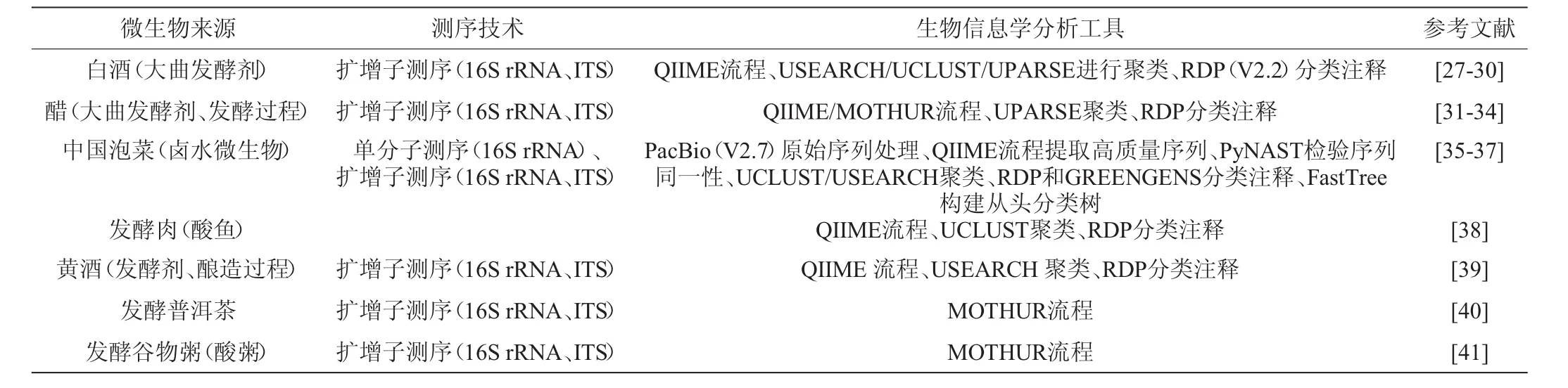
表2 基于扩增子测序分析的宏基因组数据生物信息学分析方法在传统发酵食品微生物中的代表性研究Table 2 Representative studies of bioinformatics analysis methods of metagenome data based on amplicon sequencing analysis in traditional fermented food microorganisms
由表2可以看出,现阶段用于传统发酵食品微生物研究的基于扩增子测序的核酸序列处理流程主要为QIIME和MOTHUR,聚类方法主要是USEARCH和UPARSE。而最近出现的QIIME2流程优于QIIME流程,DADA2算法也优于USEARCH、UPARSE算法。另一个比较先进的算法是UNOISE2[16],该算法经过纠正点错误获得准确扩增子序列及过滤嵌合扩增子,序列比对后聚类生成零半径分类单元(zero-radius operational taxonomic unit,ZOTU)。与DADA2不同,UNOISE2使用一次性聚类策略,该策略不使用质量分数(Q)并且只有两个预设值,能够在不同的数据集上很好的工作。ZOTU在大多数情况下优于传统的97%OTU,使用97%的同一性可能将不同序列表型不同的菌株合并成同一OTU[15,42]。
3.2 生物信息学分析方法在传统发酵食品微生物群落功能预测中的潜在应用
基于扩增子测序数据进行功能预测的生物信息学工具见表3。功能预测的常用软件有利用16S rRNA序列对细菌群落功能进行预测的分析工具FAPROTAX[17]、BugBase[18]、MMinte[19]、PICRUSt[43]及Tax4Fun[44],利用ITS序列对真菌群落功能进行预测的分析工具FUNGuild[20]等。现阶段功能预测主要集中在海洋微生物(WANG K等[45]通过PICRUSt预测得到了硫代谢相关基因的丰度信息)、肠道微生物(VRIEZE J D等[46]使用Tax4Fun对厌氧消化过程的总细菌、古菌和活跃细菌、古菌的菌群功能进行了分析,发现对活跃菌进行功能预测能够获得消化过程菌群活动和实现对消化过程机理系统层面的理解)、土壤微生物(TOJU H等[47]使用FUNGuild对ITS测序数据进行分析,揭示了土壤真菌的微共生现象)、农业微生物(LOUCA S等[48]使用FAPROTAX分析凤梨科植物微生物纤维素分解、发酵、氮呼吸等功能的菌群结构组成,发现尽管微生物群落具有稳定的功能结构,但其分类学水平的微生物群落结构具有较高的变异性,证明了相同生态系统之间的微生物群落结构变异并不意味着群落功能具有差异)研究中。然而对于微生物群落功能预测的应用,应采取谨慎的态度。一方面,由于功能预测工具都是基于一定的参考序列数据库进行功能预测,预测结果受到参考序列数据库的局限。另一方面,预测过程的数据预处理也会造成原始数据的部分损失,对预测结果产生一定的影响。同时,功能预测不能替代全基因组研究(只能对已知微生物的已知功能进行预测),但可以对后续实验设计作出指导。

表3 基于扩增子测序数据进行功能预测的生物信息学工具Table 3 Bioinformatics tools for functional prediction based on amplicon sequencing data
4 展望
目前,对于扩增子测序数据的分析已相对成熟,可供选择的各种数据库、算法、工具和平台日益增多。根据数据分析的需要,选择合适的分析工具,并对分析工具和原理进行深入的了解有利于分析过程参数调节和结果的进一步处理。随着宏基因组技术的发展,新的算法和计算平台将会不断出现。积极采用最新算法,比较不同算法之间的准确性和差异,将会加速对传统发酵食品微生物的研究。此外,为了规范传统发酵食品样本信息,有效存取海量数据信息,提供更多公用的数据源,需要建立规范的传统发酵食品微生物宏基因组信息存储平台,为宏基因组技术在传统发酵食品微生物研究中的广泛应用提供坚实的基础。
