H2S/H2硫化温度对MoP/SiO2催化剂硫醚化性能的影响
2019-06-03王立稳李风旭李明丰陈吉祥
王立稳, 李风旭, 李明丰, 褚 阳, 陈吉祥,*
(1. 天津大学化工学院 催化科学与工程系, 天津市应用催化科学与工程重点实验室, 天津 300350;2. 中国石化 石油化工科学研究院, 北京 100083)
石油化工催化材料与反应工程国家重点实验室(中国石油化工股份有限公司石油化工科学研究院)开放基金课题资助
汽油中含硫化合物燃烧产生的SO2是主要大气污染物之一,其中,绝大部分硫化物源于FCC汽油。传统脱硫工艺为加氢脱硫[1-3]。加氢脱硫工艺虽然可以有效减低汽油中硫含量,但同时会使催化裂化汽油中的烯烃过度加氢饱和,不仅降低了汽油辛烷值也增加了耗氢量。此外,催化裂化汽油中含有少量二烯烃,二烯烃在催化剂表面极易发生聚合、积炭而导致催化剂失活。为得到硫含量低、辛烷值高的催化裂化汽油,世界各国对传统加氢脱硫工艺进行了改进。其中,基于硫醚化反应与馏分切割的Prime-G+工艺可得到超低硫含量的催化裂化汽油[4]。
目前,世界范围内对硫醚化工艺方面的研究和报道较多,但对硫醚化催化剂的研究报道相对较少。研究报道的硫醚化催化剂包括金属、金属硫化物、过渡金属磷化物以及固体酸,其中,金属硫化物研究较多。肖招金等[5-7]发现,硫化后Ni/Al2O3催化剂具有良好的硫醚化性能。申志兵等[8-10]采用金属Mo改性Ni/Al2O3催化剂提高了其硫醚化性能。Huang等[11]采用Fe对Ni/Al2O3催化剂进行改性明显提高了催化剂硫醚化性能。
基于硫醚化反应机理,硫醚化催化剂应具有一定的金属性与酸性。过渡金属磷化物是一种新型加氢脱硫催化剂[12,13],兼具金属性和酸性[14,15],其表面P-OH基团为Brønsted 酸中心,而带有微量正电荷的金属位不仅是金属中心也是Lewis酸中心。因此,过渡金属磷化物作为硫醚化催化剂具有一定潜力。前期工作中,本课题组以异戊二烯及正丁硫醇的环己烷溶液为模拟油,考察了SiO2负载不同过渡金属(Mo、Ni、W、Co及Fe)磷化物的硫醚化性能[16],发现MoP/SiO2催化剂硫醚化性能最佳,正丁硫醇转化率可达100%。然而,随着反应进行,异戊二烯转化率降低;并且,反应初期,由于MoP/SiO2加氢活性较高导致深度加氢、单烯选择性较低。
为降低MoP/SiO2催化剂过度加氢性能并维持较高的异戊二烯转化率,本研究采用H2S/H2对MoP/SiO2催化剂进行硫化处理,旨在通过S改性形成相对稳定的催化剂表面结构,避免反应过程中催化剂表面被硫物种毒化发生结构变化而影响其性能。结果表明,经120 ℃硫化的MoP/SiO2催化剂具有较佳的硫醚化及选择加氢性能。
1 实验部分
1.1 试剂
环己烷(分析纯)购于天津市江天化工技术有限公司,异戊二烯(>99%)购于梯希爱(上海)化成工业发展有限公司,丁硫醇(>98%)购于阿法埃莎(Alfa Aesar)化学有限公司,正辛烷(分析纯)购于天津市光复精细化工研究所。SiO2载体购于青岛市海洋化工有限公司。体积分数3%硫化氢/氢气(体积分数3%H2S/H2)购于大连大特气体有限公司。所用氢气、氮气、氦气及CO为高纯,空气以及氨气为普纯。
1.2 催化剂的制备
首先,以SiO2负载磷酸钼为前驱体,采用程序升温还原法制备MoP/SiO2催化剂。具体过程为:配制钼酸铵((NH3)6Mo7O24·4H2O)及磷酸二氢铵(NH4H2PO4)水溶液(Mo/P物质的量比为1.0),等体积浸渍SiO2载体,然后经120 ℃干燥12 h、450 ℃焙烧4 h后制备负载磷酸钼前驱体;将1.4 g催化剂前驱体置于石英管反应器中,通入H2(320 mL/min),以10 ℃/min速率从室温升至250 ℃,再以1 ℃/min的速率升至650 ℃并恒温还原3 h制备得到MoP/SiO2。待MoP/SiO2催化剂降至室温,将气体切换为体积分数0.5% O2/N2(320 mL/min)钝化处理4 h。采用ICP-AES方法测得MoP/SiO2催化剂中Mo及P质量含量分别为17.9%及5.65%。
然后,采用体积分数3%H2S/H2硫化处理MoP/SiO2催化剂。具体过程为:取1.0 g钝化MoP/SiO2催化剂置于不锈钢固定床反应器(内径12 mm)中,在催化剂床层上下各放置2 g石英砂(20-40目);向反应器中通入H2(320 mL/min),催化剂由室温以10 ℃/min速率升至450 ℃后恒温还原1 h;还原结束后降至硫化温度,通入体积分数3% H2S/H2(100 mL/min)硫化2 h;硫化结束后,硫化后催化剂降至反应温度并切换为H2(100 mL/min)吹扫约10 h,然后在一定反应条件下评价催化剂的硫醚化性能。
未进行硫化处理的MoP/SiO2催化剂标记为MoP。在120、200、300及400 ℃硫化处理的MoP/SiO2催化剂分别标记为MoP-120S、MoP-200S、MoP-300S及MoP-400S。
1.3 催化剂的表征
采用X'Pert Pro粉末衍射仪进行X射线衍射(XRD)测试,入射线为Cu-Kα(λ=0.15418 nm),20°-80°扫描,扫描速率为8(°)/min。采用JEM-2100F场发射电子显微镜进行高分辨透射电子显微镜(HRTEM)测试。采用PHI5000VersaProbe进行X光电子能谱(XPS)测试,入射线为Al-Kα(1486.6 eV)射线,以C 1s(284.8 eV)为基准进行各元素电子结合能校正。采用VISTA-MPX电感耦合等离子发射光谱仪测定催化剂中Mo和P含量。
采用自组装设备进行NH3-TPD程序升温脱附(NH3-TPD)测试。取100 mg还原钝化的MoP/SiO2催化剂置于U型石英管反应器中,通入H2(60 mL/min)以10 ℃/min的速率升至450 ℃还原1 h;将反应器降至硫化温度通入体积分数3%H2S/H2(100 mL/min)气体硫化处理2 h;降温到100 ℃,通入NH3吸附30 min;吸附完毕后通入He(60 mL/min)吹扫催化剂表面物理吸附NH3,待TCD信号稳定后,以15 ℃/min的速率升温进行NH3-TPD,用TCD检测脱附氨,气体进入TCD前经氢氧化钠干燥。
采用自组装设备进行CO化学吸附测试。取100 mg还原钝化后的MoP/SiO2催化剂置于U型石英管反应器中,通入H2(60 mL/min)以10 ℃/min的速率升至450 ℃还原1 h;还原完毕后,降至硫化温度通入体积分数3%H2S/H2(100 mL/min)硫化2 h;然后在H2(60 mL/min)中以10 ℃/min的速率升至450 ℃,切换为He(60 mL/min)吹扫1 h;降温至30 ℃,待TCD信号稳定后用气密针向He气流中脉冲注入CO(50 μL/次),直至检测到的CO信号无明显变化。然后,根据脉冲次数和吸附CO的峰面积计算催化剂CO吸附量。
1.4 催化剂的性能评价
采用不锈钢固定床反应器(内径12 mm)评价催化剂硫醚化性能。以0.4%异戊二烯和2.8×10-4正丁硫醇(对应S含量为1×10-4)的环己烷溶液为模拟油。催化剂装填量为1 g,催化剂床层上下各放置20-40目2 g石英砂。在120 ℃、H2压力1.5 MPa、模拟油质量空速(WHSV)8 h-1以及H2/模拟油体积比为50条件下评价催化剂性能。采用北京分析仪器厂SP-3420型气相色谱仪(检测器为FID,SE-30毛细管柱(50 m×0.32 mm×3.0 μm))对液相产物进行定量分析,正辛烷为内标物。
2 结果与讨论
2.1 X射线衍射(XRD)
图1为不同催化剂的XRD谱图。由图1可知,22°处宽衍射峰归属于无定型SiO2。各催化剂XRD谱图中均有较弱的MoP衍射峰(32.17°、43.15°和67.86°,PDF#24-0771),表明MoP颗粒在载体表面分散较好,也说明即使在400 ℃硫化处理也未改变 MoP物相结构。
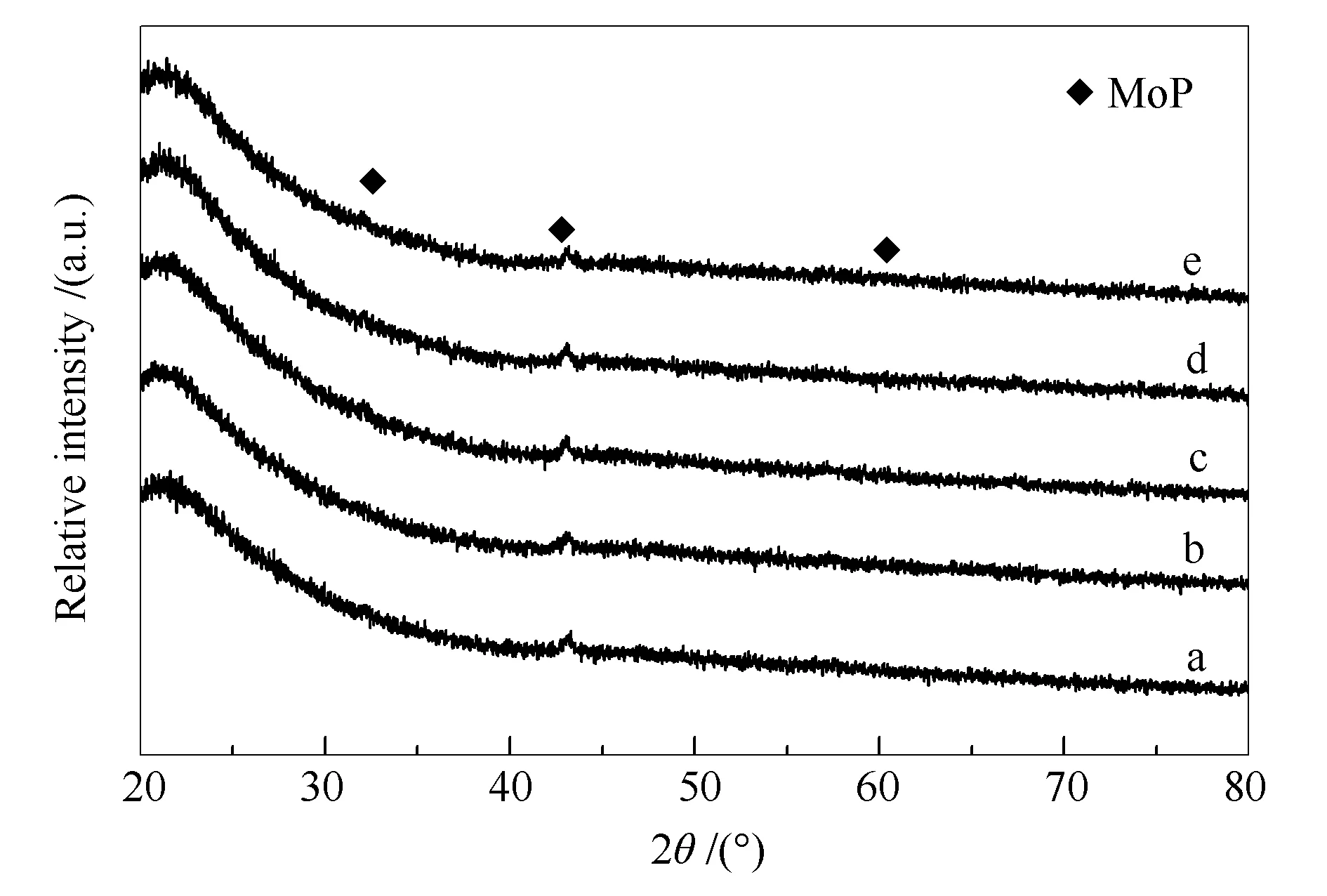
图 1 不同温度3%H2S/H2处理的MoP/SiO2催化剂的XRD谱图
2.2 高分辨透射电子显微镜(HRTEM)和能谱分析(EDX)
图2为MoP、MoP-120S和MoP-400S催化剂的HRTEM照片。各催化剂中均发现MoP晶粒(MoP(101)晶面间距均为0.21 nm)且晶粒粒径相近(约3.2 nm),表明H2S/H2硫化对MoP晶体结构未产生影响,与XRD结果一致。

图 2 MoP(a)、MoP-120S(b)与MoP-400S(c)催化剂的HRTEM照片

表 1 MoP、MoP-120S与MoP-400S中Mo∶P及Mo∶P∶S原子比
表1给示了EDX能谱分析结果。MoP-120S与MoP-400S催化剂均中均检测到S,并且MoP-120S与MoP-400S催化剂中Mo∶P原子比高于MoP催化剂。
由于硫化处理未影响MoP晶体结构,硫化后Mo∶P原子比增加可能与S原子替代催化剂表面P原子有关。XPS结果进一步证实了此点。
2.3 X光电子能谱(XPS)
为获得硫化前后催化剂表面信息,对MoP、MoP-120S和MoP-400S进行了XPS表征。图3-图5显示了催化剂中Mo 3d、P 2p以及S 2p的XPS谱图。
由图3可知,在Mo 3d区域中有三个明显的峰(分别位于228.4、231.9和235.0 eV)。Mo 3d5/2电子结合能为228.4 eV的峰归属于MoP中Moδ+物种[15,17],因其大于金属Mo的结合能(Mo0为227.4-227.8 eV[17]),因此,MoP中Moδ+物种带有微量正电荷正电荷(0<δ<+4),这与MoP中Mo向P的电子转移有关。各个催化剂Mo 3d的XPS谱图中,硫化前后MoP中Moδ+物种的Mo 3d5/2电子结合能相近,表明S未对Mo物种电子结构影响不明显。然而,如上述EDX及后续S 2pXPS结果,催化剂表面确实存在S物种,且硫化后催化剂CO吸附量明显降低(表3),说明硫物种与金属Mo确实存在一定作用。基于上述结果,推测硫与MoP中Moδ+物种之间的相互作用较弱。此外,由于催化剂进行XPS测试前进行了钝化处理,各催化剂表面还存在Mo6+物种(电子结合能分别为231.9和235.0 eV)。

图 3 MoP、MoP-120S和MoP-400S催化剂中Mo 3d的XPS谱图
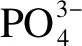

图 4 MoP、MoP-120S和MoP-400S催化剂中P 2p的XPS谱图
在S 2p的XPS谱图中(图5),MoP-400催化剂在162.7 eV处峰归属于Sδ+物种(1<δ<2)。MoP-120S催化剂谱图中S 2p峰信号不明显,可能与较低温度硫化时硫物种与催化剂相互作用较弱、表面S含量较低有关;此外,XPS测试前需对催化剂进行抽真空处理,这会导致弱吸附S物种脱附。

图 5 MoP、MoP-120S和MoP-400S催化剂中S 2p的XPS谱图
表2为硫化前后MoP催化剂表面的Mo∶P和Mo∶(P+S)原子比。

表 2 MoP、MoP-120S与MoP-400S表面原子比
由表2可知,硫化后催化剂表面Mo∶P原子比提高,而Mo∶(P+S)原子比降低,进一步说明在硫化过程中S可能替代了表面P而与钼相结合。
2.4 NH3程序升温脱附(NH3-TPD)
过渡金属磷化物具有双功能(即酸及金属功能)。金属磷化物表面P-OH基为B酸中心,而带微量正电荷的金属位为L酸中心。图6为硫化前后催化剂的NH3-TPD谱图。各催化剂NH3-TPD谱图中均有一个较宽的非对称NH3脱附峰,峰顶位于230 ℃附近,在较高温度存在肩峰。研究表明[16,19,20],低温区主脱附峰主要归属于P-OH基上NH3的脱附,而较高温度处肩峰与金属位上NH3脱附有关。由表3可知,随着H2S处理温度的提高,催化剂的酸量增加,这可能与硫物种与MoP相互作用增强、表面硫含量提高有关。硫物种可以以-SH基团存在,而-SH基团为弱酸中心[21]。

图 6 不同温度3%H2S/H2处理的MoP/SiO2催化剂的NH3-TPD谱图

表 3 不同温度H2S/H2处理的MoP/SiO2催化剂的相对酸量与CO吸附量
a: designing the acid amount of MoP as 1.00
2.5 CO化学吸附
根据报道[22,23],CO主要以线性形式吸附在过渡金属磷化物表面金属位,因此,可以用CO化学吸附法测量过渡金属磷化物催化剂表面金属位密度。如表3所示,经体积分数3%H2S/H2硫化处理后MoP/SiO2催化剂CO化学吸附量减小。随硫化温度提高,催化剂的CO化学吸附量降低。结合HRTEM-EDX与XPS结果,硫化处理后硫物种与MoP颗粒表面Mo位结合,使表面暴露的Mo原子数量减少;硫化处理温度越高,H2S与催化剂表面的Mo位相互作用越强,表面暴露的Mo位数量越少。
2.6 硫化温度对MoP/SiO2催化剂硫醚化性能的影响
异戊二烯与正丁硫醇硫醚化反应的液相产物中检测到了C4烃(丁烷、异丁烷和丁烯)、C5烃(异戊烷和异戊烯三种的异构体)、C10烃以及硫醚。在气相产物中并未检测到C1-3组分,说明反应过程中并未发生C-C键的断裂。产物中C4组分源自于正丁硫醇中C-S键断裂。C5组分为异戊二烯的加氢产物,异戊烯及异戊烷分别是异戊二烯部分及深度加氢产物。C10组分源自于异戊二烯或异戊烯聚合。催化剂表面B酸位(磷羟基与巯基)具有较强烯烃聚合能力,可以推测反应过程中会生成高沸点高聚物,但气相色谱检测未能检测出。正丁硫醇与异戊二烯或异戊烯发生硫醚化反应生成硫醚。
图7显示了异戊二烯与正丁硫醇硫醚化过程中涉及的反应,包括:正丁硫醇与异戊二烯或异戊烯发生硫醚化反应;正丁硫醇的C-S键氢解反应;异戊二烯的加氢反应;异戊二烯或单烯烃的聚合反应。

图 7 硫醚化过程中发生的反应示意图
在上述反应中,希望发生的反应为硫醚化与异戊二烯选择加氢,而应抑制C-S键氢解及异戊二烯的过度加氢和聚合反应。因此,良好的硫醚化催化剂应具备较高正丁硫醇转化率(xB)、异戊二烯转化率(xiso)、C5组分总选择性(st-C5)(尤其异戊烯选择性(sole))以及较低C4组分选择性(sC4)。
加氢、氢解反应主要发生在催化剂表面金属位上,而硫醚化与烯烃聚合主要发生在酸性位上。
2.6.1 正硫醇转化率及C4组分选择性
图8和图9为硫化温度对MoP/SiO2催化剂正丁硫醇转化率、C4组分选择性的影响。由图8与图9可知,反应12 h后各催化剂性能趋于稳定,故采用第15 h反应物转化率及产物选择性比较催化剂性能。

图 8 硫化温度对MoP/SiO2催化剂正丁硫醇转化率的影响

图 9 硫化温度对MoP/SiO2催化剂C4组分选择性的影响
由图8可知,不同温度硫化处理的MoP/SiO2催化剂上xB由高到低依次为MoP-120S>MoP≈MoP-200S≈MoP-300S≈MoP-400S。MoP-120S催化剂上xB转化率最高(93.2%)且相对稳定;随着硫化温度提高,催化剂上xB明显降低。反应12 h后,MoP-200S、MoP-300S及MoP-400S催化剂上xB稳定于73%左右。如图9所示,随反应进行,未硫化MoP催化剂上sC4逐渐降低,反应第3 h时sC4为12.8%,反应至第15 h时降为4.25%。与未硫化MoP相比,硫化MoP催化剂具有明显较低的sC4(反应至第15 h时sC4<2%)。
经研究表明[16],正丁硫醇中S-H键断裂既可以发生在催化剂表面金属位,也可发生在催化剂表面酸性位。在硫化过程中,H2S主要与MoP/SiO2催化剂表面的Moδ+物种结合。结合文献报道[22,23],随硫化温度提高,催化剂表面的Mo位数量(即CO化学吸附量)减少,催化剂加氢裂解能力降低,这不利于正丁硫醇中S-H键的断裂。NH3-TPD结果表明,随着硫化处理温度提高,催化剂表面酸量增加,但增加酸性位可能主要是酸性较弱的-SH基团,推测其酸性对正丁硫醇的S-H键裂解贡献不大。S-H键断裂主要发生在催化剂表面金属位而不是酸性位。此外,与未硫化MoP相比,硫化后MoP催化剂上sC4明显降低。C4组分由正丁硫醇经C-S键氢解生成。正丁硫醇中C-S键的键能大于S-H键[24,25],因此,C-S键比S-H键更难以断裂。硫化处理后催化剂表面暴露金属位数量降低,对S-H键与C-S键的断裂均有不利,尤其对C-S键断裂不利。总之,较高温度(200 ℃以上)硫化催化剂的C-S及S-H键断裂能力较弱,导致xB降低。
如图7所示,除了发生C-S键断裂生成C4组分,正硫醇和异戊二烯/异戊烯发生硫醚化反应,故较高xB、较低sC4意味着催化剂具有较高的硫醚化性能。各催化剂硫醚化性能高低次序如下:MoP-120S>MoP>MoP-200S≈MoP-300S≈MoP-400S。
2.6.2 异戊二烯转化率及C5组分选择性
图10为硫化温度对MoP/SiO2催化剂异戊二烯转化率的影响。由图10可知,不同温度硫化处理MoP/SiO2催化剂上xiso由高到低次序为MoP-400S>MoP-300S>MoP-200S>MoP-120S>MoP,即随着硫化温度提高,MoP催化剂上xiso增加。MoP-400S上异戊二烯转化率最高(可达99.4%)。随反应进行,硫化处理的MoP催化剂上异戊二烯转化率变化不明显或有所增加。然而,随反应进行,未硫化MoP催化剂上xiso降低(第3 hxiso约为99%,第15 h时xiso仅约为60%)。NH3-TPD和CO化学吸附结果表明,随硫化温度提高,催化剂表面的金属位数量减少、酸性位数量增加。由于金属位主要促进加氢和氢解反应,而酸性位有利于烯烃聚合。因此,硫化温度提高会促进催化剂表面发生聚合反应。虽然MoP-400S催化剂上xiso最高,但有相当一部分异戊二烯在催化剂表面发生了聚合反应,表现为C5组分选择性较低。

图 10 硫化温度对MoP/SiO2催化剂异戊二烯转化率的影响
图11显示了各催化剂上st-C5(实线)与sole(虚线)。不同温度硫化处理的MoP/SiO2催化剂上st-C5由高到低次序如下:MoP>MoP-120S>MoP-200S>MoP-300S>MoP-400S。其中,MoP催化剂上st-C5最高(82.9%左右),而MoP-400S催化剂st-C5最低(49.9%)。C5组分(异戊烯及异戊烷)是异戊二烯加氢的产物。较高st-C5意味催化剂表面异戊二烯及异戊烯聚合程度较低。总之,随硫化处理温度提高,催化剂上st-C5降低,与催化剂表面暴露金属位数量减少、酸性位数量增加有关。

图 11 硫化温度对MoP/SiO2催化剂C5组分(实线)与单烯烃选择性(虚线)的影响
图11中虚线表示各催化剂上sole。反应初期,未硫化MoP催化剂上sole较低(第3 h时仅为6.7%),说明异戊二烯发生了深度加氢;随反应进行,sole逐渐提高,反应9 h后达到81.9%(接近st-C5),这与随反应进行模拟油中正丁硫醇毒化催化剂表面金属位有关。然而,硫化处理的MoP/SiO2催化剂在反应初期sole明显高于未硫化MoP,这与硫化后催化剂表面金属位数量降低、抑制了异戊二烯深度加氢有关;并且,反应6 h后各硫化处理催化剂上sole接近st-C5。
总之,随硫化温度提高,催化剂上异戊二烯转化率及异戊烯选择性提高,但st-C5降低。综合考虑异戊二烯转化率、烯烃选择性及C5组分的总选择性,各催化剂异戊二烯加氢性能优劣次序如下:MoP-200S>MoP-120S>MoP>MoP-300S>MoP-400S。
MoP/SiO2为双功能催化剂,其表面磷羟基(P-OH)为B酸性中心,而Moδ+既为金属中心又为L酸中心。根据文献[26]可知,B酸位可催化硫醚化反应,但同时也具有烯烃聚合活性。MoP表面Moδ+具有较强的烯烃加氢反应性能,但Mo与S结合后H2的活化能力减弱、加氢活性降低。此外,金属位上活化解离的氢物种对抑制烯烃聚合也有一定作用。课题组前期工作表明[16],当异戊二烯或烯烃与B酸位H+物种结合生成了碳正离子中间体后,金属位上活性H物种会与这些碳正离子结合生成C5组分,从而抑制聚合反应的进行。因此,硫醚化催化剂表面金属与酸功能的平衡十分重要。酸性增强虽有利于硫醚化反应,但同时也促进了烯烃聚合;金属性增强会促进二烯烃深度加氢。适度硫化处理的MoP催化剂可抑制深度加氢同时可提高硫醚化性能。综合考虑正丁硫醇及异戊二烯转化率及产物分布情况,120 ℃硫化处理的MoP-120S催化剂硫醚化性能相对较佳。
3 结 论
采用体积分数3%H2S/H2硫化处理MoP/SiO2未改变MoP体相结构,但增加了催化剂表面酸量、降低了表面金属位数量;随着硫化温度的提高,催化剂表面酸量增加、金属位数量减少,这有的利于降低催化剂C-S键氢解及异戊二烯过度加氢活性、增加烯烃聚合活性。在硫醚化反应中,各催化剂正丁硫醇转化率高低次序为MoP-120S>MoP>MoP-200S≈MoP-300S≈MoP-400S,而异戊二烯选择加氢性能优劣顺序为MoP-200S>MoP-120S>MoP>MoP-300S>MoP-400S。综合考虑转化率及产物选择性,120 ℃硫化处理的MoP/SiO2性能较佳。
