棉花长期连作对新疆农田土壤古菌群落演替的影响
2019-05-31张伟杜钰
张伟,杜钰
新疆特殊环境物种保护与调控生物学实验室/新疆师范大学生命科学学院,新疆 乌鲁木齐 830054
古菌(Archaea)也叫古细菌(Archaebacteria),是自然界三大生物类群之一(Martin,2005)。古菌可能是最古老的生命体,早先常被发现生活于各种极端自然环境下,如大洋底部的高压热溢口、热泉、盐碱湖等地(Martin,2005)。近年的研究表明,土壤中氨氧化是氮素转化过程的第一步也是限速环节,而氨氧化古菌在农田氨氧化过程中起着重要促进作用(Hatzenpichler,2012)。由于大多数古菌都难以被培养和分离,有关其在土壤碳(C)、氮(N)、硫(S)、铁(Fe)等元素循环过程中所起的作用,以及环境治理潜在的开发前景鲜见报道(包丽君等,2017)。
新疆是中国最早种植棉花的地区之一,也是目前中国唯一的长绒棉种植基地,棉花长期连作在当地是极为普遍的现象(倪天麒等,2002)。由于新疆地区位于中亚腹地且远离海洋,其土壤和气候具有相当特殊性。在当地种植棉花需要采用地膜保墒、人工定期灌溉、大量施用氮肥和各种农药等人为干预措施(金建新,2008)。研究表明,这些农作措施极大地改变了当地土壤环境(李腾等,2018),然而更多的研究是从土壤养分、理化性质等角度进行分析(王树林等,2017),而有关棉花连作对古菌群落结构的影响,以及它们的改变与棉花连作障碍和土传病害的发生、发展之间的关联性鲜有报道(陈一峰等,2015;Gloor et al.,2010)。
虽然传统微生物研究手段难以深入开展土壤古菌群落结构研究,但近些年发展起来的非培养技术手段为此类研究提供了契机。目前,高通量测序技术得到快速发展,不仅测序长度不断增加,测序通量飞速提高,而且能够用于对比分析的古菌数据量也不断增大,目前该技术已成为最强有力的研究微生物生态系统的工具(李锐等,2015;Lazarevic et al.,2009)。本研究采用该方法对比分析了新疆地区原生态及长达 30年棉花连作过程中土壤古菌群落结构组成和差异,有助于从土壤微生物生态的角度解析当地干旱盐碱土壤环境下古菌群落结构特征,以及土壤古菌群落结构对棉花长期连作、其他作物轮作和与之配套的农作措施产生的响应。
1 材料和方法
1.1 样品的采集
采样时间、方式和位置:2015年6月5日(此时棉花枯萎病和黄萎病开始发作)于新疆阿瓦提县棉田,依据当地农技部门提供的信息,分别选择棉花连作 0(未开垦土壤)、1、3、5、10、15、20、25、30 a的棉田和长年连作棉田改种玉米1年的农田为研究对象,所有样地(2500亩,约为166.67 hm2)集中在新疆阿瓦提县,采 样 范 围 在 79°45′81″- 79°46′55″E , 39°31′47″-39°45′50″N 之间。采用“五点”取样法采集土壤样品,在每一连作年限样地的地头、地中、地脚各随机选择 5株棉花或玉米,用内径为6 cm的土钻在距植株5 cm处垂直打下,取1-20 cm深度土壤。将15个相同连作年限土壤合并为1个土样,共计10个土样。按连作年数依次编号为 AWTG0、AWTG1、AWTG3、AWTG5、AWTG10、AWTG15、AWTG20、AWTG25、AWTG30和AWTGL。土样采集后于-4 ℃保存,带回实验室,一周内完成DNA提取、测序。
1.2 DNA提取、PCR扩增和高通量测序(DNA extraction,PCR and pyrosequencing)
土壤样品总DNA的提取使用FastDNA® spin kit(MP bio,Santa Ana,USA),操作方法按照试剂盒说明,每个样品做 3个重复。各重复样品总DNA按照相同的量合并后储存于-80 ℃下冰箱中,备用。使用PARCH519R(CAGCCGCCGCGGTAA)/ARC915R(GTGCTCCCCCGCCAATTCCT)引物来扩增各样品总DNA的16S rRNA基因V4+V5片断(Ovreas et al.,1997;Muyzer et al.,1993)。30 μL 反应体系:Phusion Master Mix(2×)15 μL,上下引物各 1.5 μL(2 μM),各样品 DNA(1 ng·μL-1)10 μL,双蒸水 2 μL。Bio-rad T100梯度 PCR 仪,反应程序:98 ℃,1 min;98 ℃,10 s;50 ℃,30 s;72 ℃,30 s,共3O个循环;72 ℃,5 min后稳定在4 ℃。紫外分光光度计(NanoDrop-1000,USA)准确定量扩增获得的各样品16S rRNA基因519-934区片段,各样品等浓度充分混匀后,在1×TAE、2%琼脂糖胶中电泳后,使用 Thermo Scientific公司 GeneJET胶回收试剂盒回收 280-450 bp的主带。上述样品使用New England Biolabs公司的 NEB Next® Ultra™ DNA Library Prep Kit for Illumina建库试剂盒进行文库的构建,构建好的文库经过Qubit定量和文库检测,合格后于Illumina HiSeq平台上机测序。本文所测得的序列在 NCBI SRA数据库中的登录号为SRS1624762。
1.3 生物信息和统计分析(Bioinformatics and statistical analysis)
Novogene公司自编脚本被用于截去Barcode和引物序列,每个样品上游引物序列前端有6 bp的特征序列用于区别不同的样品,各样品优化后可供精准分析的序列至少40000条以上(Magoc et al.,2011)。UPARSE pipeline(v7.0.1001)被用于对获得的各样品序列进行OTU等相关生物信息分析,相似度设置为97%。对OTUs代表序列进行物种注释,用RDP Classifier(Version 2.2,http://sourceforge.net/projects/rdp-classifier/)方法与 GreenGene数据库(http://greengenes.lbl.gov/cgi-bin/nph-index.cgi)进行物种注释分析(设定阈值为0.8-1),并分别在各个分类水平:kingdom(界)、phylum(门)、class(纲)、order(目)、family(科)、genus(属)、species(种)统计各样本的群落组成。
运用Qiime软件(Version 1.7.0)分析各样品的αDiversity并计算 Observed-species、Chao1、Shannon、Simpson、ACE、Goods-coverage指数,运用R软件(Version 2.15.3)绘制稀释曲线、Rank abundance曲线、物种累积曲线、韦恩图;并进行Alpha多样性指数组间差异分析(Caporaso et al.,2010)。在β Diversity分析中,运用Qiime(Version 1.7.0)软件计算weighted和unweighted Unifrac距离并构建UPGMA样品聚类树。运用R语言vegan包vegdist和hclust进行样品相似度聚类、OUT热图和 β多样性距离计算及热图(PCA、PCoA和NMDS等图)绘制,其中PCA分析使用R软件的ade4包和ggplot2软件包,PCoA分析使用R软件的WGCNA,stats和ggplot2软件包,NMDS分析使用R软件的vegan软件包(Wang et al.,2007)。
2 结果与分析
2.1 各样品古菌群体数据
10个样品中可用于精准分析的序列共有626590条,平均长度为383 bp,其中AWTG30样品隶属于古菌界的序列数最少,为45937条。各样品获得的有效序列数达到40000条时,可检测到种的饱和度曲线已接近平台期,测序错误率小于 1%碱基比率(Q20)和 0.1%碱基比率(Q30)分别达98.34%和 96.6%以上,所有样品覆盖度介于 0.992-0.997之间(表1)。从界到种水平上,各样品获得的能用于准确分类的序列数量比例较接近,但不能被准确分类的序列数随着分类精度的提高不断增多,其中在门分类水平最多只有 2.05%(AWTG25)的序列不能被分类(图1A),而在属水平上不能被明确分类的序列最多达到了 66.7%(AWTG15)(图1B)。
2.2 α多样性指数及OTU
各菌群多样性指数中,AWTG0样品 Shannon指数最低为4.76,其次是AWTG30样品,为5.335,最高的是AWTGL样品,为6.269,且各棉花连作样品 Shannon指数在连作过程中表现出反复波动(表 1)。各样品 Simpson、Chao1和 ACE指数与Shannon指数变化结果比较一致,最低的都是AWTG0,但Simpson指数最高的是AWTG10样品,为 0.945,Chao1和 ACE指数最高的都是样品AWTG25,分别为1604.915和1711.09。这3个指数在连作过程中也表现出反复波动,但波动的幅度和频率各不相同(表1)。可归类的OTU数量中,AWTG0样品最低为 763,其他各样品 OTU数在1091(AWTG30)-1626(AWTG25)之间,AWTGL样品介于中间水平,为1605(图2)。对比分析各样品OTU差异,其中所有样品共有的OTU有232个。各样品特有OTU中,原生态样品为80个,各连作样品特有 OTU数介于 14(AWTG30)-125(AWTG25)之间,而所有样品中 AWTGL最多,为127个(图3)。
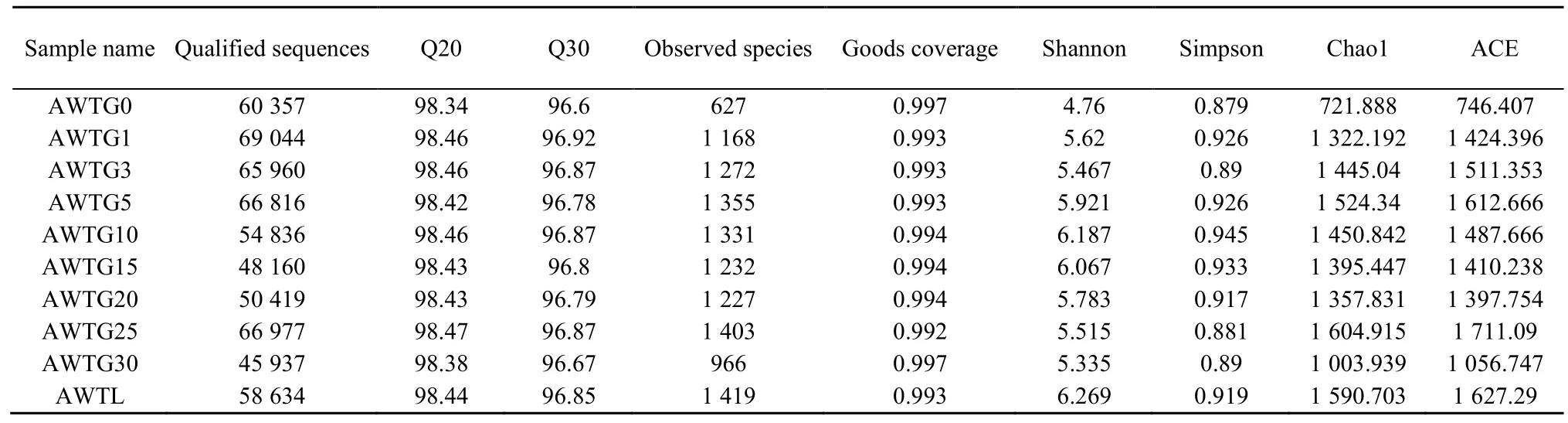
表1 10个不同连作年限土样古菌群落群体数据及多样性指数Table 1 Diversity of the 10 soil samples in archaea with different succession cropping histories
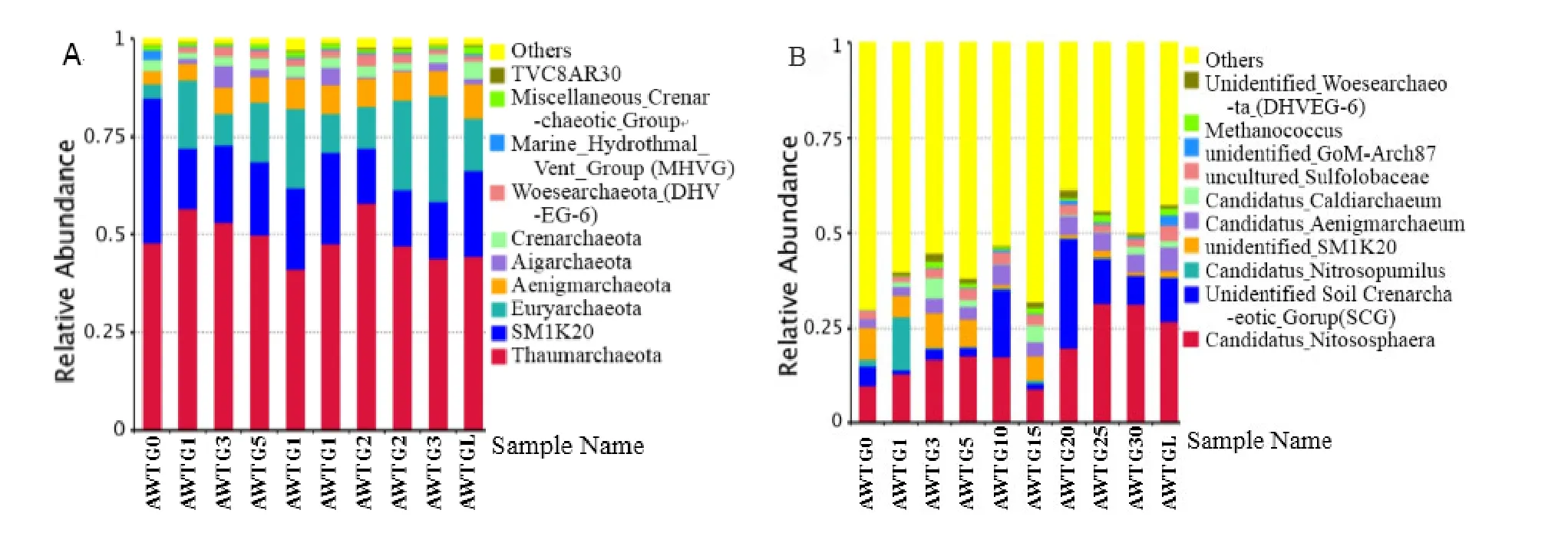
图1 各样品中古菌丰度最大的前10个门(A)和属(B)占比Fig. 1 Abundances of different phylums (A) and genus (B) in archaea, the abundance is presented in terms of percentage in total effective archaea sequences in the 10 soil samples
2.3 原生态土壤古菌群落结构特征及对棉花连作后产生的响应
在门分类水平上,当地原生态土壤古菌群落主要由Thaumarchaeota(47.91%)、SM1K20(37.07%)、Euryarchaeota(3.51%)、Aenigmarchaeota(3.32%)、Crenarchaeota(2.78%)和 Marine_Hydrothermal_Vent_Group(MHVG)(2.32%)等6个丰度占比较大的门组成(总占比为 96.93%)。棉花长期连作过程中Thaumarchaeota和Crenarchaeota的丰度无明显变化,SM1K20的丰度出现大幅度下降(平均为15.82%),Euryarchaeota的丰度出现大幅度上升(平均为16.44%),Aenigmarchaeota的丰度增长了1倍,Marine_Hydrothermal_Vent_Group(MHVG)的丰度减少为原来的1/11(图1A)。在属分类水平上,当地原生态土壤古菌群落结构中有 66.23%以上尚不能被准确分类,其余的主要由Candidatus_Nitrososphaera(9.74%)、unidentified_SM1K20(8.46%)、unidentified_Soil_Crenarchaeotic_Group(SCG)(5.16%)、Candidatus_Aenigmarchaeum(2.24%)、uncultured_Sulfolobaceae(2.16%)和Candidatus_Nitrosopumilus(1.78%)等6个丰度占比较大的属组成;而棉花连作后Candidatus_Nitrososphaera和Candidatus_Aenigmarchaeum的丰度增加近1倍,unidentified_SM1K20和uncultured_Sulfolobaceae的丰度无明显变化,unidentified_Soil_Crenarchaeotic_Group(SCG)的丰度依据连作年限不同出现巨大波动,Candidatus_Nitrosopumilus的丰度减少了近8倍(图1B)。玉米轮作1年使得丰度占比最大的10个属中Sulfolobaceae和GoM-Arch87的丰度成倍增加(图1B)。
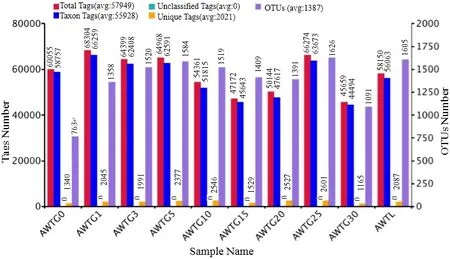
图2 各样品中古菌测得的序列数据统计信息及其OTU数Fig. 2 Total tags, taxon tags, unclassified tags, unique tags and OTUs of the 10 samples
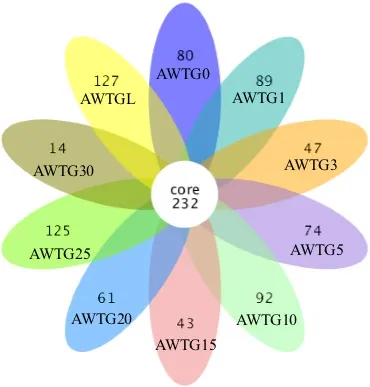
图3 各样品中特有及共有OUT数Fig. 3 Venn diagram of unique number of OTUs in the 10 soil samples
2.4 样品间古菌群体结构β多样性比较
unweighted unifrac聚类结果显示原生态样品单独为一族,而棉花连作的样品聚在了一大族,其中又以连作年限长短不同明显分为2族,轮作样品被聚在长期连作族(图4A)。weighted unifrac聚类结果明显不同,该聚类方法是所有样品按连作年限长短直接将其分为2族,其中原生态样品和轮作样品被聚在长期连作族,但原生态样品与其他连作样品差异较大,而轮作样品与长期连作样品差异很小(图4B)。主坐标分析(PCoA)的unweighted数据显示,原生态样品与其他样品距离较远,而棉花连作年限长和短的又相对各成为一个群体分别位于横轴的上下方,且轮作样品与连作年限长的样品较集中(图5A);weighted数据显示原生态样品与轮作样品距离较近,而棉花连作年限长和短的又相对各成为一个群体,分别位于第一、三象限(图5B)。非度量多维尺度结果(NMDS)显示,原生态样品与所有连作样品间距离非常大,而且连作年限长的样品和轮作样品相对集中且几乎全部重叠,连作年限短的样品相对集中且几乎全部重叠(图6)。
3 讨论
3.1 新疆地区土壤古菌资源及其与环境的适应性
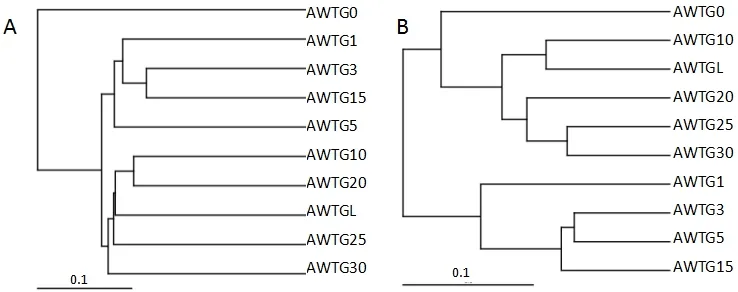
图4 各样品古菌群落结构相似度聚类Fig. 4 Analysis of sample similarity in the 10 soil samples
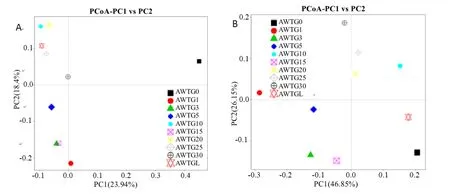
图5 各样品古菌群落结构主坐标分析Fig. 5 Principal Coordinate Analysis (PCoA) of 10 soil samples calculated with QIIME
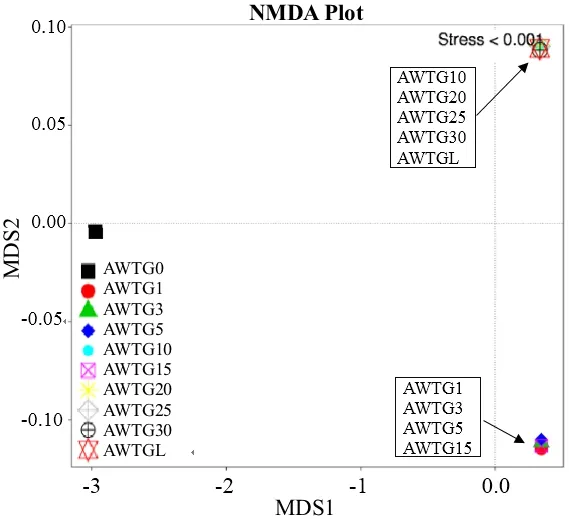
图6 各样品NMDS plots分析Fig. 6 Nonmetric Multidimensional Scaling (NMDS) analysis of the 10 soil samples
古菌群落结构特征与其生存环境密不可分,可作为特定地质环境的标志物(曹鹏等,2012)。新疆地处中亚腹地,气候干旱少雨,土壤盐渍化极严重,被开垦作为棉田的土地多分布在绿洲和沙漠之间的过渡带,植被稀疏、土壤有机质含量极少,且富K、少P、缺N,pH值平均8.52以上,诸如此类土壤环境下的古菌群落结构组成研究少有报道(韩春丽等,2010)。通过对当地原生态土壤环境样品(AWTG0)序列数据分析可知,其中的古菌门类群多达14个。丰度最大的 Thaumarchaeota(奇古菌门)在该环境中占比 47.91%,而已有关于 Thaumarchaeota的报道认为其具有氧化氨获得能量的功能(沈菊培等,2011;陈玉连等,2017;Noah et al.,2003),这与当地土壤理化特性不符,因此 Thaumarchaeota在当地土壤环境中的功能有待深入研究。目前有关Euryarchaeota和Crenarchaeota门古菌研究报道相对较多,并认为她们是海洋水环境和沉积物中的优势种群,当中包含许多产甲烷菌、耐盐的盐杆菌、嗜热菌等(张丽梅等,2012),但本研究发现这两个门的古菌在当地原生态土壤环境中分别也占有 3.51%和2.78%的丰度,其中原因值得深入研究。SM1K20门多为尚未分离鉴定的古菌(Durbin et al.,2012),其功能研究报道也微乎其微,而其丰度在该环境中占比达37.1%,处于第二位。被认为是由小的古菌细胞组成的Aenigmarchaeota门(范习贝等,2017)在该环境中的丰度也达到了3.32%。此外,该原生环境中丰度较高的门还有 Marine_HydrothermaVl_ ent_Group(MHVG)(2.32%),Miscellaneous_Crenarchaeotic_Group(1.36%)。推测该土壤环境虽然较为苛刻,但孕育的特有未知古菌资源较为丰富,对其在当地土壤生态中的功能展开深入研究有助于挖掘该环境下的古菌资源。
3.2 当地土壤古菌群落对棉花连作产生的响应
众所周知,土壤是最复杂的微生态系统之一,气候条件、土壤理化性质等都是影响土著微生物群落结构组成的重要因素(Pimental et al.,1992)。新疆棉区由于作物种类单一等因素,在一个小区域内同时存在连作不同年限(0-30 a)的棉田,这给研究古菌群落适应土壤的开垦和棉花连作的响应提供了很大的便利。首先,所有耕作样品α和β多样性指数变化规律都表明土壤开垦是导致当地土壤古菌多样性上升的直接因素,而且耕作的第一年升高幅度最大。分析认为定期灌溉、施氮肥等农业管理措施极大地改变了当地土壤理化性质(Zhang et al.,2013),致使被检测到的古菌种属的数量和丰度都有较大幅度的提高。其次,所有样品形成一种按连作年限长短各自聚类和调整幅度逐渐趋小的走势,并在棉花多年连作后最终形成新的相对稳定的古菌群落结构。推测土壤古菌群落结构的这种变化过程和当地半年耕作半年休田的栽培方式有一定关联,因为土壤古菌群落组成可以在每年棉花春夏耕作期间发生适应性调整,而在冬秋休田期间进行部分恢复。第三,本研究主要在门分类水平对比各样品间不同类群丰度和幅度的变化规律,发现了耕作对古菌群落结构造成的影响。至于棉花连作过程中的变化规律则需要在较低分类水平上进行对比分析,但由于可以用来准确对比并定位这些序列所代表的菌属及其功能的信息有限,因此不适合深度解析其关联性。对于能被准确分类的序列,依然可以分析其随连作年限延长而表现出的规律性变化。比如,在纲分类水平,Aenigmarchaeota_Incertae_Sedis是连作之后丰度才增加的。在目分类水平,Sulfolobales的丰度在耕作前后均无明显变化。在属分类水平,Candidatus_Nitrososphaera非常明确地表现出随连作年限延长而丰度递增的趋势,这和该属具有氨氧化功能(李明等,2017)及因当地针对土壤理化特性而采取的以施氮肥为主的措施比较相符。另外,栽培作物改为玉米后,引起个别古菌属的组成丰度发生较大调整,其中的关联和轮作能降低棉花病虫害的具体原因都值得深入研究。
4 结论
(1)新疆阿瓦提县原生态土壤含有丰富的特有古菌资源。当地土壤的开垦和棉花的种植极大地改变了古菌群落结构组成并提高了其多样性。棉花的长期连作促使形成了新的古菌群落结构组成。棉花和玉米轮作能快速调整一些古菌属的丰度,研究其关系有助于更好地阐释作物与古菌间的互作机制。
