黄酒熟麦曲中细菌多样性的评价
2019-05-31崔梦君折米娜张振东赵慧君郭壮
崔梦君,折米娜,张振东,赵慧君,郭壮
(湖北文理学院食品科学技术学院 鄂西北传统发酵食品研究所,湖北 襄阳 441053)
作为黄酒酿造中的重要原料,麦曲中包含霉菌、酵母菌和细菌等微生物,由于微生物与生物环境的互生、共生、寄生和拮抗等关系,逐渐形成了一个复杂且具有多样性的微生物区系,同时对黄酒风味也起到了至关重要的作用[1-2]。谭婷婷等从北方黄酒麦曲中分离出5 株霉菌和1 株酵母菌,分别为多枝横梗霉、米曲霉、广紫青霉、杂色曲霉、交链孢霉和酿酒酵母[3],曹珏等发现米根霉、微小毛霉、米曲霉和烟曲霉是绍兴黄酒麦曲中的主要真菌[4]。目前关于黄酒发酵过程中挥发性风味物质的变化以及真菌群落结构进行了大量研究[5-7],然而关于麦曲中细菌多样性的研究却鲜有报道。
变性梯度凝胶电泳(denatured gradient gel electrophoresis,DGGE)是一种能够快速对环境中的微生物群落进行评估的分子生物学技术[8],被广泛应用于食醋[9]、泡菜[10]、香肠[11]、大酱[12]和黄酒[13]等发酵食品的微生物多样性解析中。Illumina MiSeq 是一种高通量测序方法,可以以相对较低的成本产出大量序列,与其他二代高通量技术相比增加了微生物群落分析的测序深度,同时降低了试验成本,使其成为根据大规模标记基因测序研究不同环境中微生物群落的新方法[14-15],目前在农业饲料[16]、发酵食品[17]和环境卫生[18]微生物解析领域均有广泛的应用。
本研究以黄酒麦曲为研究对象,在提取样品微生物宏基因组DNA 的基础上,利用DGGE 与MiSeq 高通量测序技术相结合的手段对其细菌多样性进行评价,同时结合生物信息学手段对其微生物群落结构进行解析,以期为后续麦曲中微生物资源的挖掘提供理论支持。
1 材料与方法
1.1 材料与仪器
熟麦曲:采集自浙江省丽水市,编号分别为MQ1、MQ2、MQ3 和 MQ4;三羟甲基氨基甲烷、乙酸、乙二胺四乙酸、丙烯酰胺、甲叉双丙烯酰胺、去离子甲酰胺、尿素、过硫酸铵、四甲基乙二胺、乙醇、冰醋酸、甲醛、硝酸银、氢氧化钠:国药集团化学试剂有限公司;D5625-01 DNA 提取试剂盒、DNA marker、聚合酶链式反应(polymerase chain reaction,PCR)清洁试剂盒:京科博汇智生物科技发展有限公司;2PCR×mix:南京诺唯赞生物科技有限公司;rTaq、2'-脱氧核苷酸-5'-三磷酸混合物(dNTP MIX)、pMD18-T vector:大连宝生物技术有限公司;引物:武汉天一辉远生物科技有限公司。
引物信息见表1。
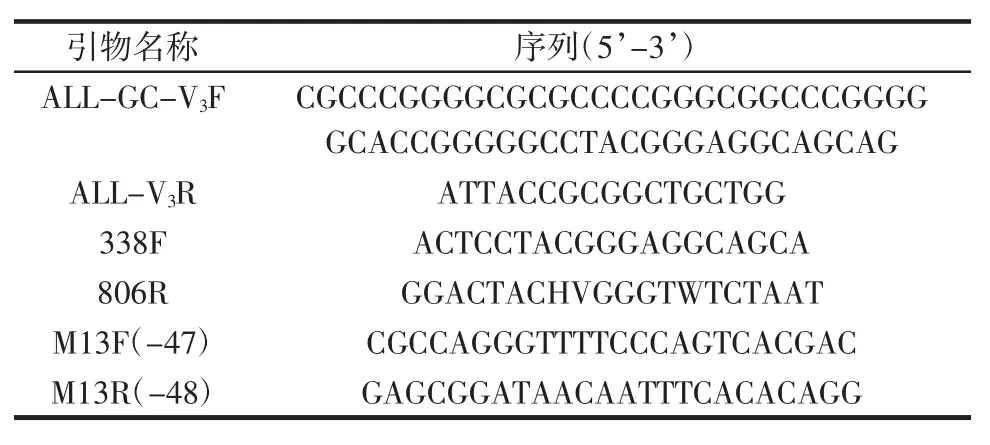
表1 引物信息Table 1 The information of primers
VeritiTM96-well Thermal Cycler PCR 仪:美国AB公司;NanoDrop 2000:美国 Thermo Fisher 公司;DCodeTMSystem:美国 Bio-Rad 公司;DYY-12 电泳仪:北京六一仪器厂;MiSeq PE300 高通量测序平台:美国Illumina 公司;R920 机架式服务器:美国DELL 公司;CT15RE 冷冻离心机:日本HITACHI 公司;Bio-5000 plus 扫描仪:上海中晶科技有限公司。
1.2 方法
1.2.1 样品微生物宏基因组提取与检测
采用D5625-01 试剂盒提取麦曲微生物宏基因组DNA,用0.8%琼脂糖凝胶进行电泳检测并使用NanoDrop 测定浓度。
1.2.2 DGGE 电泳及优势条带测序
将样品总DNA 作为模板进行聚合酶链式反应(PCR),采用细菌通用正向引物ALL-GC-V3F 和反向引物ALL-V3R 对样品16S rDNA V3 区域进行扩增。PCR 扩增总体积为 25 μL,包括 10×PCR Buffer(含Mg2+)2.5 μL,dNTP 2 μL,正、反向引物各 0.5 μL,rTaq 0.5 μL,模板 1 μL,无菌超纯水补至 25 μL。PCR 扩增程序:95℃预变性 4 min,95℃变性 30 s,55℃退火30 s,72℃延伸 30 s,循环 30 次,最后 72℃终延伸10 min。所得扩增产物用2%的琼脂糖凝胶电泳检测。使用Bio-Rad 公司Dcode 突变检测系统对上述PCR产物进行电泳,采用8%的丙烯酰胺凝胶,变性梯度为35%~52%。DGGE 条件:恒温 60℃,0.5 TAE 电泳缓冲液,上样量 10 μL,电压先 120 V,运行 80 min,后电压80 V,运行780 min。电泳结束后,采用硝酸银法染色,使用扫描仪将电泳图进行拍照,找出各样品特征条带并回收胶块。用不带GC 夹板的引物(ALL-V3F 和ALL-V3R)将回收胶块再次进行PCR 扩增。试剂盒纯化PCR 产物并进行载体连接和克隆培养,挑取阳性克隆子送往武汉天一辉远生物科技进行测序。
1.2.3 麦曲细菌16S rRNA PCR 扩增及MiSeq 高通量测序
参考蔡宏宇等方法进行样品细菌16S rRNA PCR扩增及MiSeq 高通量测序[19]。扩增体系总体积为20 μL,包括 5×PCR 缓冲液 4 μL,dNTP mix 2 μL,带有 7个核苷酸标签(barecode)的正向引物338F 和反向引物806R 各 0.8 μL,rTaq 酶 0.4 μL,模板 10 ng,无菌超纯水补至 20 μL。扩增程序为:95℃预变性 3 min,95℃变性 30 s,55℃退火 30 s,72℃延伸 45 s,循环 30 次,72℃终延伸10 min。用1.0 %琼脂糖凝胶电泳检测PCR 扩增产物,寄至上海美吉生物医药科技进行Illumina MiSeq PE300 平台进行高通量测序。
1.2.4 序列拼接及质量控制
高通量测序数据成功下机后,参考董蕴等方法对序列进行质控和拼接[20]:根据成对序列之间的重叠关系,将双端序列拼接成一条序列并去除不合格序列,从而完成序列校正。利用QIIME 分析软件,将相似度〉97%以上的序列归类为一个操作分类单元(operational taxonomic unit,OTU),从而确定各序列对应微生物的分类学地位及相对含量,同时根据Chao 1 指数和Shannon指数对各样品的微生物丰富度和多样性进行分析。本研究将在4个样品中均存在的OTU 定义为核心OTU。
1.3 数据处理
使用Origin 8.5 软件对稀释曲线及香农指数曲线作图,同时对麦曲样品中优势细菌属进行柱状图的绘制。各特征条带序列系统发育树由Bio Edit 软件和MEGA 7.0 软件共同绘制。基于OTU 水平的Venn 图由在线网站(http://bioinfogp.cnb.csic.es/tools/venny/index.html)进行绘制。
2 结果与分析
2.1 麦曲细菌DGGE电泳图谱及条带测序分析
本试验首先利用PCR-DGGE 技术对麦曲样品中的细菌多样性进行了电泳分析,麦曲中细菌PCRDGGE 图谱结果如图1所示。

图1 麦曲中细菌PCR-DGGE 图谱Fig.1 PCR-DGGE analysis of bacteria in wheat Qu
由图1可知,共有8个条带在电泳图中明显表现出来。条带 2、3、4、5、6 和 7 均存在于麦曲样品中,而条带1 仅存在于MQ4中,条带8 存在于MQ1 和MQ4中,由此可知各样品细菌组成存在差异。将各条带切胶回收进行PCR 扩增、测序、比对及系统发育树绘制,其结果如表2所示。
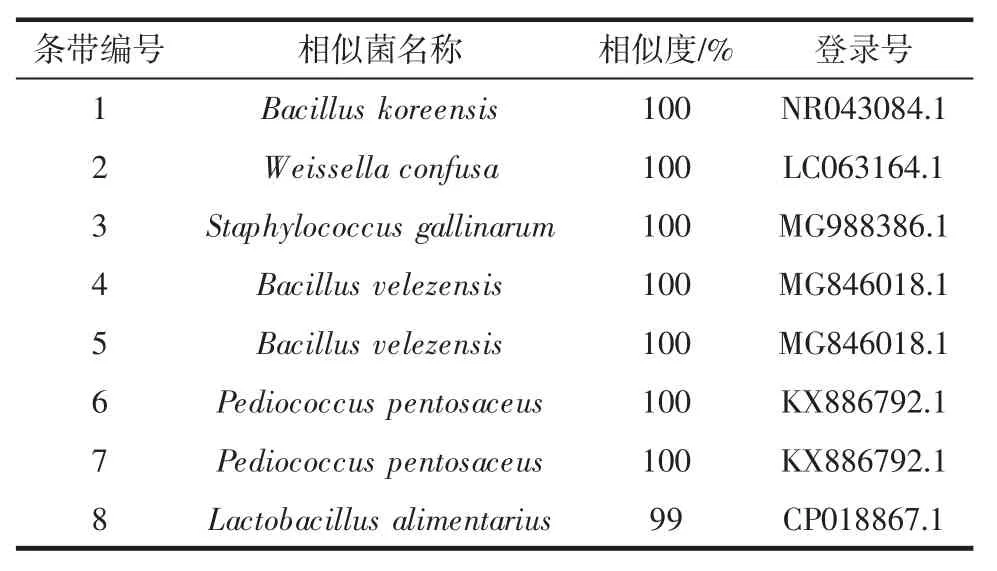
表2 麦曲DGGE 指纹图谱中条带比对结果Table 2 The blast results of bands in DGGE fingerprint of wheat Qu
由表2可知,条带 1、2 和 3 分别与 Bacillus koreensis(韩国芽孢杆菌)、Weissella confusa(融合魏斯氏菌)和Staphylococcus gallinarum(鸡葡萄球菌)相似度达 100%,条带 4 和 5 与菌 Bacillus velezensis(贝莱斯芽孢杆)相似度达100%,同时条带6 和7 与Pediococcus pentosaceus(戊糖片球菌)相似度达100%,而条带8 与Lactobacillus alimentarius(食品乳杆菌)相似度为99%。系统发育树见图2。
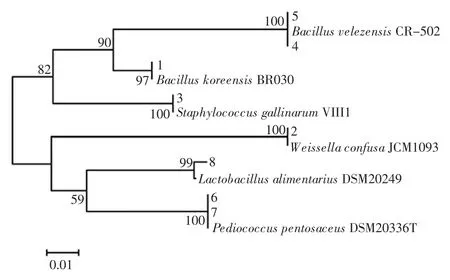
图2 系统发育树Fig.2 Phylogenetic tree
由图2可知,系统发育树被分为两大分支,说明各分支上聚集的菌株与数据库中的模式菌有较强的亲缘关系。其中条带 1、3、4 和 5 聚为一类,而条带 2、6、7和8 聚为一类。在DGGE 指纹图谱中不同条带代表不同的细菌种类,体现了样品微生物的丰富程度,条带越亮或越粗表示其代表的种群生物量越大[21]。由此可见,麦曲中的细菌主要隶属于Bacillus(芽孢杆菌)、Weissella(魏斯氏菌)和Pediococcus(片球菌)。薛景波利用16S rDNA 分子学方法对黄酒接种生麦曲发酵过程中微生物群落的变化研究发现Weissella confusa(融合魏斯氏菌)和Enterobacter cloacae(阿氏肠杆菌)的丰度较高,并被确定为优势细菌[22]。
2.2 序列丰富度及多样性分析
本试验进一步将各麦曲样品进行MiSeq 高通量测序,4个样品共产生220 707 条序列。根据两步UCLUST法对所有序列进行分析,首先经100%相似度进行聚类,共得到73 688 条代表性序列,依据97%相似度聚类后得到5 726个OTU,平均每个样品1 431个OTU。样品16S rRNA 测序情况及各分类地位数量如表3所示。

表3 样品16S rRNA 测序情况及各分类地位数量Table 3 16S rRNA read counts and the number of identifiable units on different taxonomical levels
由表3 亦可知,MQ1 样品中细菌群落丰富度最大,而MQ2 样品中细菌群落多样性最高。稀释曲线可以直接反映测序数据量的合理性,香农曲线是用来衡量个样品之间微生物群落的差异性[23]。本研究进一步通过稀疏曲线和香农指数曲线对测序深度是否满足生物信息学分析进行了评价,其结果如图3所示。

图3 稀疏曲线图和香农曲线图Fig.3 Rarefaction curve and Shannon index curve
由图3可知,随着测序序列数的增加,虽然会有新的细菌种系型可能会被发现,但是微生物多样性却处于饱和状态,由此可知上述测序量可以满足生物信息学分析。
2.3 基于不同分类学地位细菌群落相对含量分析
纳入本研究的4个麦曲样品,共产生220 707 条序列,通过同源性比对将其鉴定为12个门、29个纲、49个目、96个科和170个属,只有0.94%和5.64%的序列不能鉴定到门和属水平。在门水平上,Firmicutes(硬壁菌门)、Proteobacteria(变形菌门)和Actinobacteria(放线菌门)为优势细菌门,其中Firmicutes(硬壁菌门)平均相对含量最高,而Actinobacteria(放线菌门)平均相对含量最低,分别为87.97%和0.94%,由此可知隶属于Firmicutes(硬壁菌门)的细菌为麦曲样品中的优势细菌。同时各样品中硬Firmicutes(壁菌门)相对含量分别为 88.13%、89.13%、90.04 和 84.48%,Proteobacteria(变形菌门)相对含量分别为9.17%、8.93%、7.18%和10.72 %,而Actinobacteria(放线菌门)相对含量为1.13%、0.57%、1.34%和1.58%,由此可知虽然上述各细菌门均存在于各样品中,但是相对含量却存在差异性。本研究进一步将相对含量〉1%的细菌属进行分析,结果如图4所示。
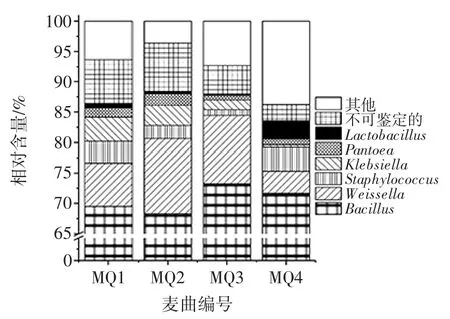
图4 麦曲样品中优势细菌门相对含量分析Fig.4 Analysis of relative abundance of dominant bacterial in wheat Qu samples at the phylum level
由图4可知,优势细菌属分别为Bacillus(芽孢杆菌属)、Weissella(魏斯氏属)、Staphylococcus(葡萄球菌)、Klebsiella(克雷伯菌属)、Pantoea(泛菌属)和Lactobacillus(乳杆菌属),其平均相对含量分别为70.70%、8.55 %、2.67 %、2.29 %、1.32 %和 1.07 %。利用 Illumina MiSeq 测序平台,刘芸雅对绍兴黄酒麦曲及黄酒发酵过程中细菌多样性进行了分析,结果发现麦曲中优势菌为Bacillus(芽孢杆菌属)和Saccharomyces(糖多孢菌属),同时Bacillus(芽孢杆菌属)、Staphylococcus(葡萄球菌属)和Lactobacillus(乳杆菌属)为黄酒发酵过程中的优势细菌属[24]。利用传统微生物学方法,张中华对熟麦曲中的细菌进行了培养、分离和鉴定,结果发现 Bacillus(芽孢杆菌属)、Staphylococcus(葡萄球菌属)和Pantoea(泛菌属)细菌存在于熟麦曲中[25]。任清通过纯培养和分子生物学方法对北宗黄酒麦曲中的细菌进行鉴定,结果发现Bacillus(芽孢杆菌属)为麦曲中的优势细菌[26],该报道与本研究结论一致。
本研究进一步将每个样品中的OTU 数量及序列进行统计,结果如图5所示。
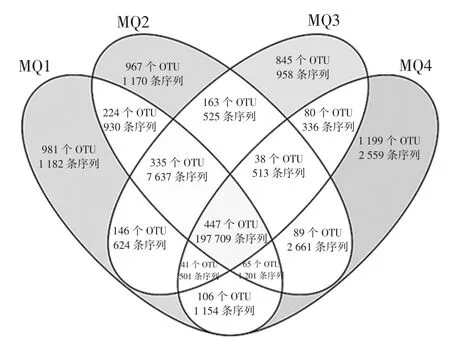
图5 基于OTU 水平的Venn 图Fig.5 Venn diagram based on OTU level
由图5可知,4个样品中共发现5 726个OTU,而仅存在1个样品中的OTU 为3 992个,占OTU 总数的69.72%,序列数为5 869 条。同时出现2 次和3 次的OTU 分别为808个和479个,分别占 OTU 总数的14.11%和8.37%,包含序列数分别为6 230 条和9 852条。值得一提的是在4个样品中共发现447个核心OTU,占OTU 总数的7.80%,包含197 709 条序列。进一步分析发现,在核心OTU中有7个OTU 的平均相对含量〉1%,结果如图6所示。
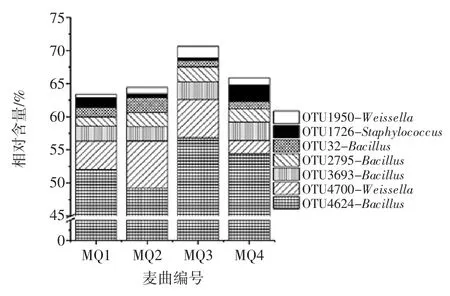
图6 平均相对含量>1%核心OTU 比较分析Fig.6 Comparative analysis of average relative abundance more than 1%of core OTU
由图6可知,平均相对含量〉1%的核心OTU 分别为OTU4624、OTU4700、OTU3693、OTU2795、OTU32、OTU1726 和OTU1950,其平均相对含量分别为54.44%、1.97%、2.78%、1.99%、1.10%、2.56%和1.03%。进一步将上述7个核心OTU 序列进行Blast 比对发现OTU4624、OTU2795 和 OTU3693 隶属于 Bacillus(芽孢杆菌属),OTU4700 和 OTU1950 隶属于 Weissella(魏斯氏属),而OTU 隶属于Staphylococcus(葡萄球菌属)。
3 结论
本研究以麦曲为研究对象,采用PCR-DGGE 与MiSeq 高通量测序技术相结合的手段对其细菌多样性进行评价分析。结果发现,麦曲中的细菌组成具有较高的多样性,主要为硬壁菌门、变形菌门和放线菌门,其中隶属于硬壁菌门的芽孢杆菌属与魏斯氏属是为优势菌属。通过本试验的开展,为后续麦曲中微生物资源的挖掘提供了理论支持。
