κ-卡拉胶比例对明胶凝胶体系凝胶特性、水分分布及微观结构的影响
2019-05-23郭琦王欣刘宝林
郭琦,王欣,刘宝林
(上海理工大学 医疗器械与食品学院,上海,200093)
明胶是一种食品工业中广泛使用的凝胶剂,具有良好的胶凝性、热可逆性和亲水性,以及“入口即化”的特性[1],但明胶单独使用时,有凝胶时间较长、凝胶强度弱、形成凝胶所需浓度较高等局限性。将明胶与其他食用胶复配可改善其凝胶特性,扩大胶体适用范围,满足不同加工要求。研究表明,卡拉胶与明胶复配可形成凝胶强度和持水性均较好的凝胶体,表现出良好的协同增效作用。周爱梅等[2]研究发现,当明胶/卡拉胶比例为1∶3时,可形成良好凝胶,在两者比例为5∶3时凝胶强度达到最大。王晓婧[3]研究发现,随卡拉胶比例的增加,卡拉胶-明胶混合凝胶的硬度增加,弹性相对减弱,而持水性先增强后降低。SINTHUSAMRAN等[4]对鱼明胶凝胶特性的研究发现,添加卡拉胶可使鱼明胶凝胶体系的凝胶强度增加,但过高的卡拉胶含量则会削弱混合凝胶体系的弹性。
凝胶体系的微观结构、水分状态及分布与其宏观性具有密切关系。THRIMAWAWITJANA等[5]研究了KCl对κ-卡拉胶的机械性能,微观结构和流变性质的影响,发现加入低浓度KCl时,可增加体系的交联密度,聚合物排列类似于蜂窝结构,这有助于凝胶体系具有较高的拉伸性能。黄远芬等[6]运用低场核磁共振技术(low-field nuclear magnetic resonance,LF-NMR)研究了明胶-蔗糖/NaCl凝胶体系中水分状态及比例的变化,发现体系的弛豫特性与凝胶强度间有良好的相关性。田海娟等[7]研究发现,大豆分离蛋白中11S球蛋白比例对蛋白凝胶水分分布、凝胶特性及微观结构有显著影响。刘施琳等[8]研究了多个因素对琼脂凝胶强度及弛豫特性的影响,发现琼脂凝胶的弛豫时间T23与凝胶强度存在极显著(P<0.01)负相关,即凝胶中水分流动性越小,凝胶强度越大。
基于以上分析,本文以明胶-κ-卡拉胶体系为研究对象,采用质地剖面(texture profile analysis,TPA)/穿刺分析、低场核磁场共振(LF-NMR)、计算机断层扫描技术(computed tomography,CT)和流变仪重点研究κ-卡拉胶比例对明胶凝胶强度、质构特性、水分分布、微观结构和动态黏弹性的影响,分析2种胶的相互作用特点,为明胶与κ-卡拉胶形成的复配胶在食品中的应用提供理论支持。
1 材料与方法
1.1 材料与仪器
κ-卡拉胶(食品级),上海康达食品工程有限公司;B型猪骨明胶(食品级),上海联合食品添加剂有限公司;去离子水(自制)。
分析天平(PWC254型),艾德姆衡器(武汉)有限公司;恒温磁力搅拌器(85-2A型),金坛市科析仪器有限公司;水浴恒温振荡器(THZ-82型),常州智博瑞仪器制造有限公司;低场核磁共振分析仪(PQ-001型,氢谱,磁场强度(0.5±0.08)T,共振频率23 MHz),上海纽迈电子科技有限公司;质构分析仪(TA.XT.Plus Texture Analyser),英国Stable Micro System公司;台式真空冷冻干燥机(LGJ-10D型),北京四环科学仪器厂;工业CT机(XT H 225型),尼康仪器(上海)有限公司;流变仪(Discovery HR-3),美国TA仪器公司。
1.2 试验方法
1.2.1 明胶-κ-卡拉胶混合体系的配制
在预实验的基础上,称取一定量的κ-卡拉胶和明胶置于烧杯中,加入去离子水,配制成总胶质量浓度为3.0 g/100mL的均匀溶液,使κ-卡拉胶占总胶比例分别为0、10%、20%、30%、40%、50%、60%、70%、80%、90%、100%,室温2 h充分溶胀后,80 ℃恒温振荡30 min,形成均匀的明胶-κ-卡拉胶混合溶液,准确移取3 mL溶液于直径为15 mm的核磁共振专用试管中,10 mL溶液至10 mL的烧杯中,120 mL溶液至冻力瓶中,4 ℃下放置12 h备用。测试前将样品温度恢复到室温。
1.2.2 凝胶强度的测定
参照黄远芬[6]的方法,采用质构仪测量样品的凝胶强度,具体参数如下:探头P/0.5,测试前速度1.50 mm/s,触发力10 g,测试速度1.00 mm/s,测试距离20.00 mm,返回速度10.00 mm/s。当探针进入凝胶深度达4 mm时,测得的最大力即为样品的凝胶强度。
1.2.3 质构特性的测定
在PROMMAJAL[9]方法的基础上略作修改,测量参数如下:P/36R的探头;测试前速度:1.50 mm/s;触发力:5 g;测试速度:1.00 mm/s;压缩比:70%;返回速度:10.00 mm/s,根据质地剖面的力度-时间曲线,得到样品的硬度、弹性、咀嚼性、内聚性、黏性和回复性等的信息。
1.2.4 水分分布特性
应用LF-NMR分析凝胶体系的水分分布特性。在黄远芬等[6]的基础上略加修改。具体参数如下:CPMG脉冲序列,重复采样等待时间:15 000 ms,重复扫描次数:4次,谱宽:100 kHz,半回波时间:250 μs,回波个数:18 000个,采样点数:3 750 126。
对采样得到的CPMG指数衰减曲线应用T-invfit软件进行反演拟合,得到样品的多组分弛豫图谱,并获知各弛豫峰的弛豫时间(按出峰时间从小到大命名为T21、T22…T2i等)及归一化后的峰面积百分比(S21、S22…S2i等)。当将样品看作一个整体组分分析时,可反演得到样品的单组份弛豫时间(T2W,ms)。
1.2.5 凝胶微观结构
应用工业CT对冻干后的凝胶样品进行微观结构分析。具体过程如下:将凝胶样品切分为2 mm×2 mm×2 mm的正方体,在-45 ℃预冻180 min后,于-10 ℃,压力<10 Pa,一次干燥400 min,再在4 ℃,压力<10 Pa,二次干燥400 min,获得凝胶冻干样品,密封备用。CT扫描参数:管电压70 kV,管电流80 μA,拍摄张数1 000张,进行扫描,得到初步图像,经去噪得到CT扫描图像,进行三维重构,然后选取重构图中的代表性切面进行分析。
1.2.6 动态黏弹性的测定
参照刘国军等[10]的方法,应用旋转型流变仪(DHR-3, TA, USA)进行样品的动态黏弹性分析。温度由Peltier板和恒温循环水浴装置共同控制。取1 mL配制好的样品加到已预热至70 ℃的样品台上,用蒸馏水密封防蒸罩以防止实验样品水分过度挥发造成测量误差。采用小幅振荡模式,固定测量间距(Gap=1 mm),频率为1 Hz,温度由70 ℃降至15 ℃后再升至70 ℃,变温速率为5 ℃/min,测定降温和升温过程中G′(储藏模量)和G″(损耗模量)的变化。冷却过程中G′和G″的交汇点的温度为胶凝温度;加热过程中G″和G′的交汇点的温度为熔化温度。
1.3 数据分析方法
试验均设3次平行,每个样品重复检测3次,应用SPSS 18.0分析凝胶强度、质构特性与LF-NMR弛豫特性的相关性,应用ANOVA进行方差分析和Duncan多重检验,以P<0.05作为差异显著性判断标准,应用Origin 8.0软件进行绘图。
2 结果与分析
2.1 凝胶强度
凝胶强度能够揭示凝胶网络结构形成的数量及紧密程度,可以用来评估食品的加工处理方法及其对产品质量的影响。
由图1-A可知,不同κ-卡拉胶比例的混合凝胶的力度-时间曲线差异明显,随κ-卡拉胶比例增大,曲线的应力逐渐增大,且破裂强度越大、破裂点距离越大,探头压迫凝胶使其破裂时的压强和凝胶破裂时探头行进的距离(破裂点距离)均相对增大。说明凝胶体系较难破裂且胶体的延展性较好。由图1-B可以看出,单纯明胶凝胶的凝胶强度最低,仅为(36.91±5.17)g,而单纯κ-卡拉胶所形成的凝胶具有最高的凝胶强度(670.80±34.94)g,二者之间差异显著(P<0.05),当体系中添加10%的κ-卡拉胶后,其凝胶强度显著升高,且混合凝胶体系的凝胶强度与κ-卡拉胶比例间呈良好的二项式关系(R2=0.966 9),说明κ-卡拉胶比例越高,其对混合体系凝胶强度的影响越大,在较高的κ-卡拉胶比例下,形成以κ-卡拉胶为主体的网状结构。这是由于κ-卡拉胶中带负电荷的硫酸盐基团与明胶中带正电荷的基团通过局部静电相互作用形成一定的网络结构[11],随κ-卡拉胶比例的增加,二者间的交互作用加强,分子间的联结程度增加,使凝胶网络结构更加致密,表现为凝胶强度的逐渐增大。
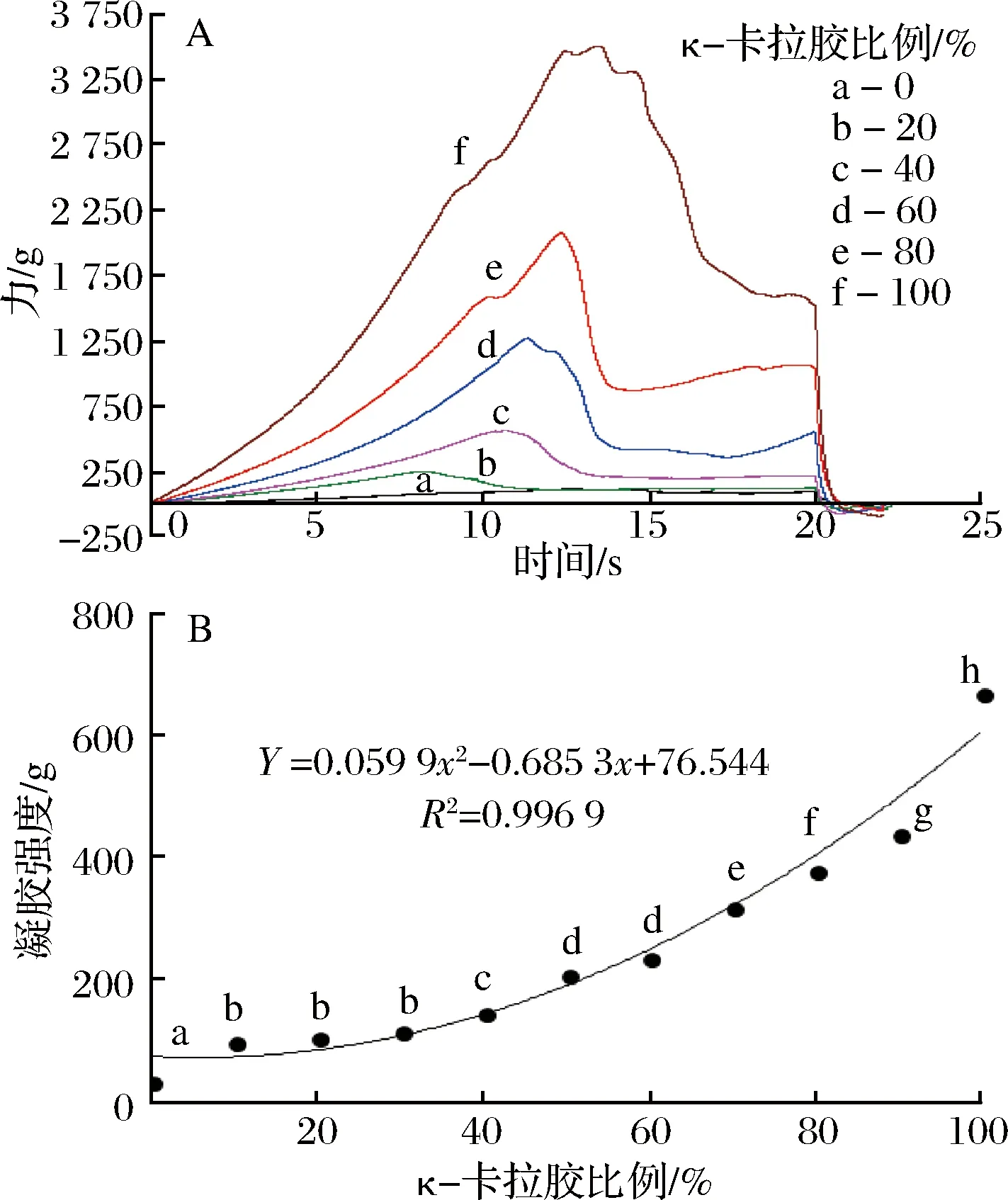
图1 部分混合凝胶体系的穿刺实验力度-时间曲线(A)、凝胶强度(B)随κ-卡拉胶比例的变化Fig.1 Change in intensity-time curves (A) and the gelstrength (B) of some mixed gel system with carrageenan ratio注:A中字母表示卡拉胶不同比例;B中不同字母表示差异显著(P<0.05)。下同。
2.2 TPA质构特性
质地剖面分析(TPA)是一种易于量化的分析技术,已被广泛应用于食品工业[12]。部分混合凝胶压缩测试得到的力度-时间曲线如图2所示。
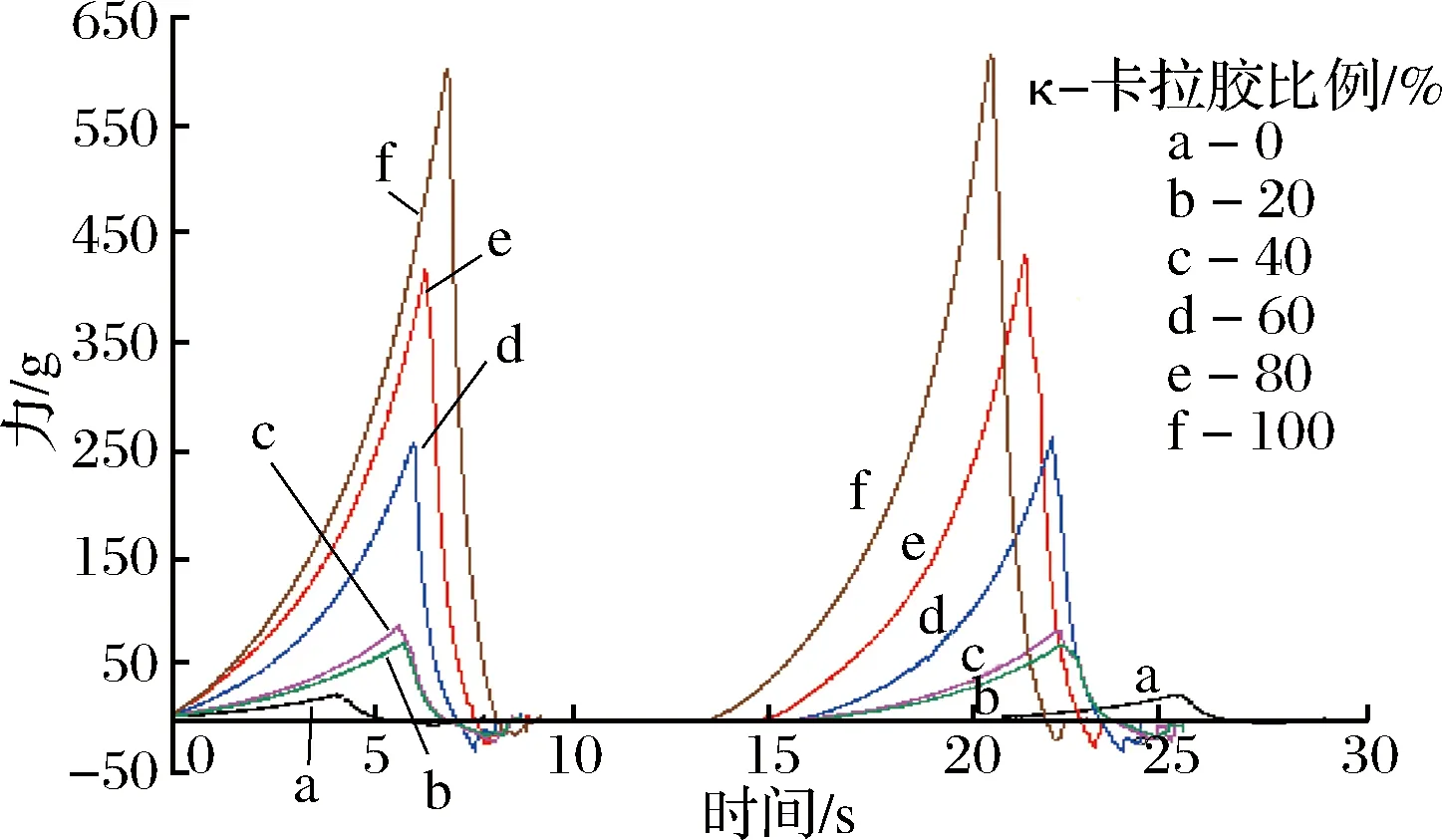
图2 部分混合凝胶体系TPA实验的力度-时间曲线Fig.2 Changes in velocity-time curve in TPA experimentof some mixed gel system
由图2可知,不同κ-卡拉胶比例的混合凝胶的力度-时间曲线差异明显,随κ-卡拉胶比例增大,曲线的力值逐渐增大。尽管不同比例κ-卡拉胶的凝胶峰值力度不同,但随着探针移入样品,所有凝胶的峰值在短时间内均迅速地增加。经过曲线计算可以得到食品的质构特性,如硬度、弹性、咀嚼性、内聚性、回复性及黏性等参数。
2.2.1 混合凝胶体系的硬度与弹性
混合凝胶体系的硬度、弹性随κ-卡拉胶比例的变化如图3所示。
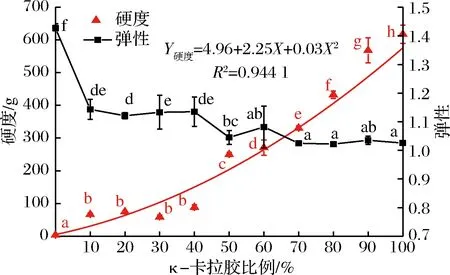
图3 混合凝胶体系的硬度、弹性随κ-卡拉胶比例的变化Fig.3 Changes in hardness, springiness of mixed gelsystem with carrageenan ratio
由图3可知,单纯明胶体系的硬度仅为(3.41±0.82)g,而单纯κ-卡拉胶凝胶硬度高达(617±27.87)g。添加κ-卡拉胶后,混合凝胶体系的硬度显著升高,且随κ-卡拉胶的增大,其硬度呈现良好的二项式增大的趋势(R2=0.944 1),尤其当κ-卡拉胶比例大于50%时,混合凝胶的硬度变化幅度加大。弹性的变化则有所不同,明胶凝胶体系的弹性为(1.43±0.01),而添加κ-卡拉胶后,混合凝胶体系的弹性显著减小(P<0.05),κ-卡拉胶比例为10%时,体系的弹性降至1.02,继续增大κ-卡拉胶比例对体系弹性的影响较小,当κ-卡拉胶比例大于50%时,体系的弹性又显著降低。说明κ-卡拉胶的添加使体系在外力去除后恢复到变形前状态的程度相对减小。HUANG等[13]研究卡拉胶浓度对大米淀粉凝胶质构的影响时发现,卡拉胶凝胶通常弹性较小,在压缩测试过程中容易破碎。SINTHUSAMRAN等[14]对含有不同比例κ-卡拉胶的混合鱼凝胶的物理和流变性质的研究中也发现,添加卡拉胶会使明胶凝胶的弹性降低,更易碎,在卡拉胶比例较高时尤其明显。
2.2.2 混合凝胶体系的咀嚼性与内聚性
混合凝胶体系的咀嚼性、内聚性随κ-卡拉胶比例的变化如图4所示。
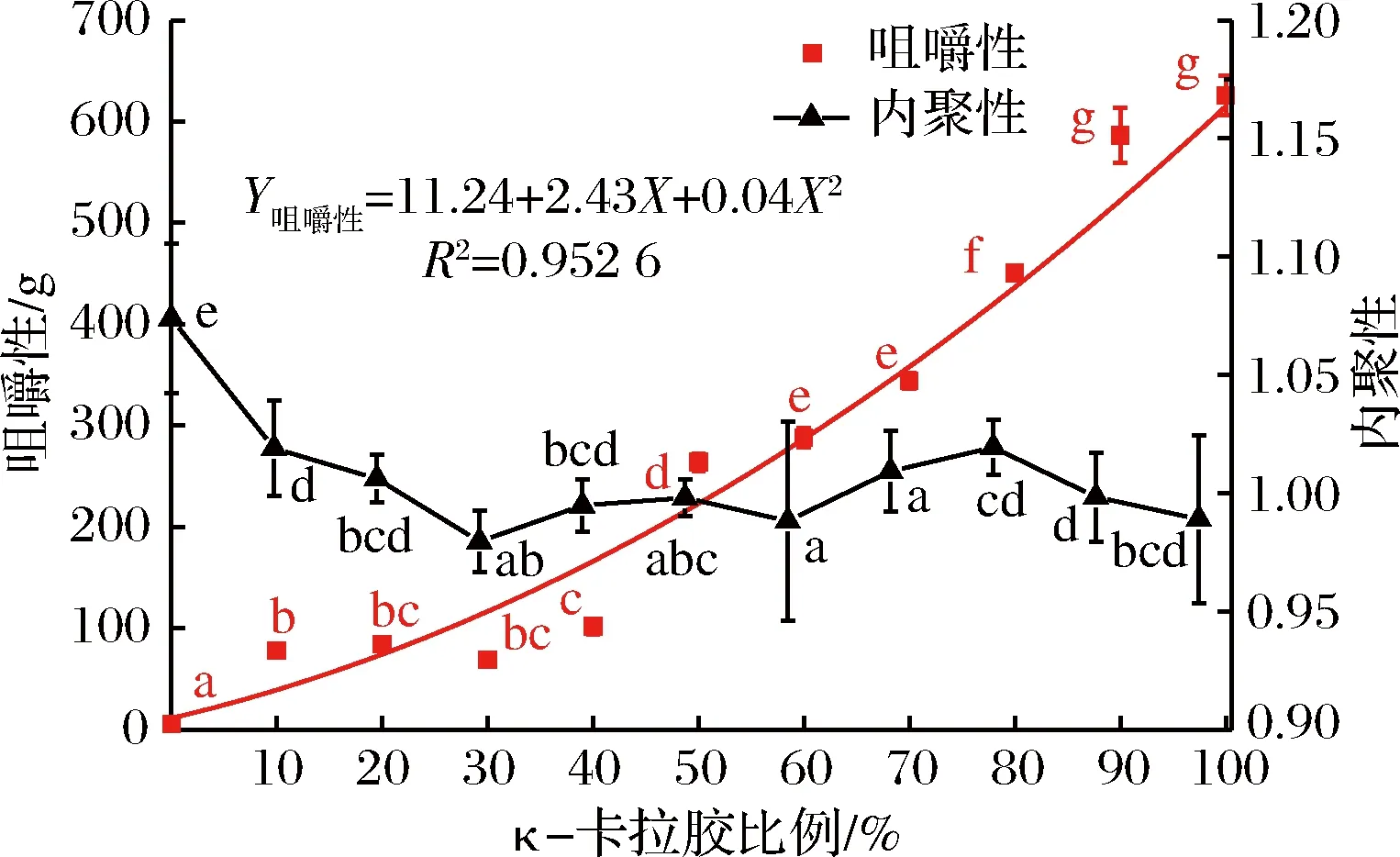
图4 混合凝胶体系的咀嚼性、内聚性随κ-卡拉胶比例的变化情况Fig.4 Changes in chewing and cohesiveness of mixed gel system with carrageenan ratio
图4表明,当混合体系中含有10%的κ-卡拉胶时,其咀嚼性从(5.25±1.38)g显著增加至(77.53±2.91)g,随着κ-卡拉胶比例的增加,咀嚼性呈二项式显著上升趋势(P<0.01,R2=0.952 6),尤其是κ-卡拉胶比例大于50%后,咀嚼性的增幅进一步增加。咀嚼性与硬度密切相关,硬度越大,咀嚼性越高。
内聚性是指形成样品形态所需内部结合力的大小。从内聚性的变化来看,添加κ-卡拉胶后,混合凝胶的内聚性发生显著性变化(P<0.01),尤其是当κ-卡拉胶比例<30%时,变化更为显著,当κ-卡拉胶比例>30%时,体系内聚性相对稳定。说明添加κ-卡拉胶后,凝胶体系内部分子间的结合力相对减弱,形成的凝胶抵抗受损、保持凝胶完整性的能力相对降低,这与κ-卡拉胶的凝胶特性相符,其成胶相对较脆,易碎。
2.2.3 混合凝胶体系的回复性与黏性
混合凝胶体系的回复性、黏性随κ-卡拉胶比例的变化如图5所示。
从图5可以看出,随着κ-卡拉胶比例的增大,混合凝胶的黏性和回复性呈现先下降后上升的趋势,在κ-卡拉胶比例达到30%时达到最低。但随κ-卡拉胶比例的变化,混合凝胶的回复性变化范围较小,从0.18降低到0.11。
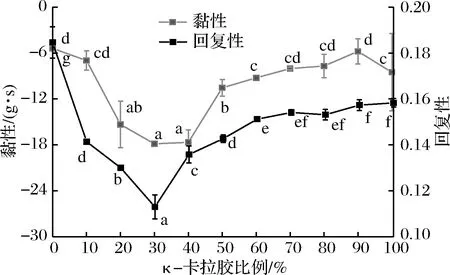
图5 混合凝胶体系的黏性、回复性随κ-卡拉胶比例的变化情况Fig.5 Changes in adhesiveness and resilience of mixed gel system with carrageenan ratio
2.3 水分分布特性
低场核磁共振的自旋-自旋弛豫时间(T2)信息可以表征具有不同流动性的水分子在体系内的分布[15],部分明胶-κ-卡拉胶凝胶体系的LF-NMRT2多组分弛豫图谱如图6所示。
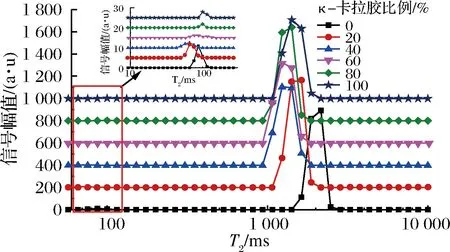
图6 部分混合凝胶体系的LF-NMR多组分弛豫图谱随κ-卡拉胶比例的变化情况Fig.6 Changes in LF-NMR multi-component relaxation map of partially mixed gel system with carrageenan ratio
由图6可知,在T2多组分弛豫图谱上,明胶、κ-卡拉胶及κ-卡拉胶-明胶混合凝胶体系均出现了2个弛豫峰,T21位于50.94~121.14 ms,T22位于911.13~1 438.70 ms,说明凝胶样品中的氢质子分为了弛豫速率有所区别的两类。其中,凝胶体系中的结合水与κ-卡拉胶或明胶基团结合紧密,受到的束缚力较大,弛豫速率大,弛豫时间较短,与T21对应;而T22则代表了束缚于凝胶网络中的不易流动水中的氢质子的响应。当体系中κ-卡拉胶比例≤50%时,随其比例的增加,弛豫图谱整体左移,弛豫时间相对减小,而当κ-卡拉胶比例>50%后,T21则相对增大,而T22相对稳定。
明胶-κ-卡拉胶凝胶体系的弛豫时间(T2w、T21、T22)和峰面积比例(S21、S22)随κ-卡拉胶比例的不同而变化,结果如图7所示。
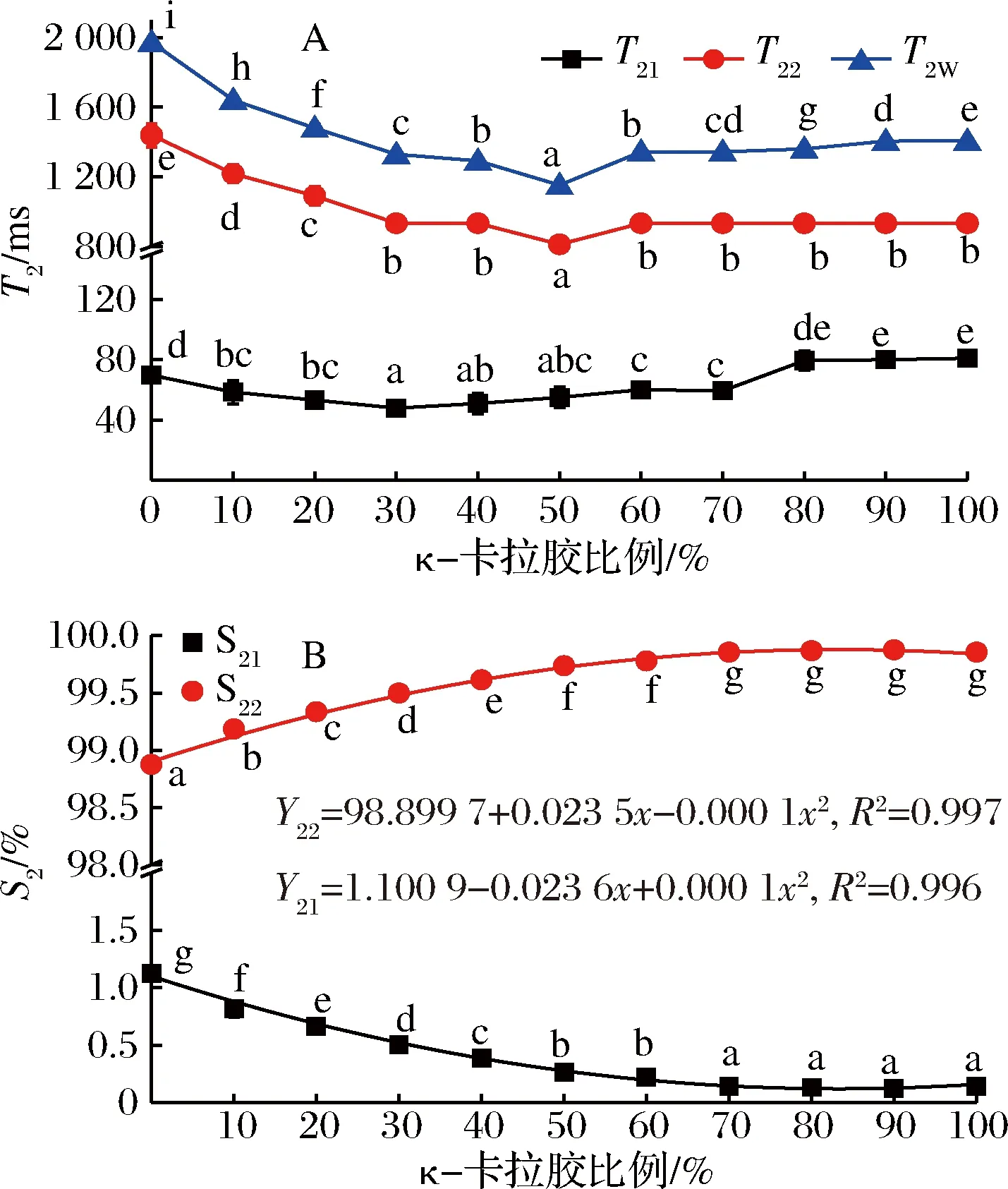
图7 混合凝胶体系的弛豫时间(A)和峰面积比例(B)随κ-卡拉胶比例的变化Fig.7 Changes in relaxation time(A) and peak area ratio of mixed gel system with carrageenan ratio
单组份弛豫时间(T2W)是被测样品所含氢质子弛豫信息的平均值,可反映样品整体的弛豫特性。由图7-A可知,当体系中κ-卡拉胶比例≤50%时,随其比例的增加,混合凝胶体系的T2W显著减小,而κ-卡拉胶比例>50%后,T2W则相对增大,但变化幅度较小。这是由于在2种胶体的相互作用下,体系整体的分子间作用力相对增强,氢质子自由度降低,平均的弛豫速率相对增大,表现为弛豫时间的缩短[16]。T22的变化与T2W相似,当体系中κ-卡拉胶的比例小于50%时,T22从1 438.70 ms逐渐降低至811.13 ms,说明κ-卡拉胶的加入使凝胶网络的束水能力增强,不易流动水中氢质子的流动性进一步降低,当κ-卡拉胶比例大于50%后,凝胶体系中不易流动水的流动性相对稳定,T22基本不变。而代表混合凝胶中结合水的T21整体变化幅度不大,仅在小范围内波动,说明结合水的流动性比较稳定,受κ-卡拉胶比例变化的影响较小。
与弛豫时间相比,峰面积比例的变化更有规律性。由图7-B可知,随κ-卡拉胶比例的增大,S21呈二项式下降趋势(R2=0.996),而S22呈二项式增加趋势(R2=0.997)。TANG等[17]对水分在原生未加工的淀粉颗粒中的分布和动态研究发现,核磁弛豫图谱的相对峰面积与其对应的水分分数成比例。随κ-卡拉胶比例的增加,混合凝胶的网络状结构更为成熟且致密,使网络对水的束缚能力增强,表现为S22的相对增大。显著性分析表明,当κ-卡拉胶比例大于70%后,S21、S22相对稳定,继续增大κ-卡拉胶比例对于水分分布无明显影响。
2.4 凝胶强度、质构特性与LF-NMR弛豫特性的相关性
运用相关性分析对质构特性、凝胶强度与LF-NMR弛豫特性进行相关性分析,结果如表1所示。
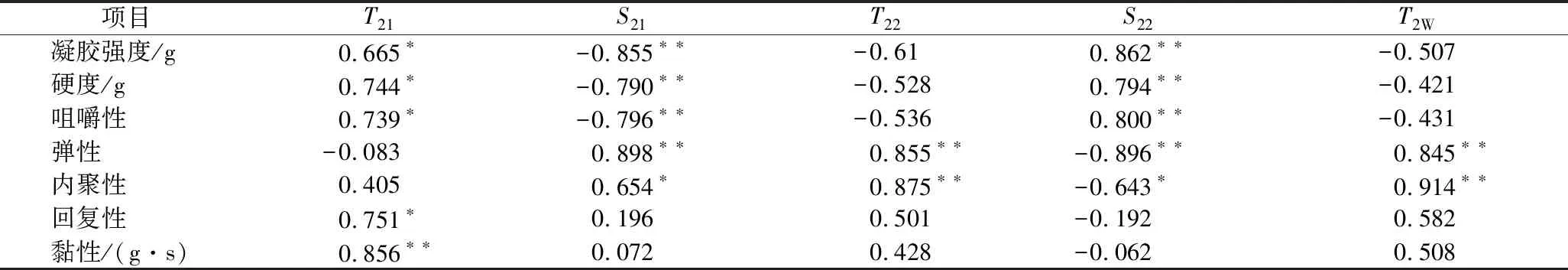
表1 混合凝胶体系的凝胶强度、质构特性与LF-NMR弛豫特性的相关性分析Table 1 Correlation analysis of gel strength, texture characteristics content with LF-NMR relaxation characteristics
注:** 在 0.01 水平(双侧)上显著相关;* 在 0.05 水平(双侧)上显著相关。
由表1可知,凝胶强度、质构特性参数与多组分弛豫特性参数均呈较好的相关性。其中,凝胶强度、硬度、咀嚼性与S21呈极显著负相关(P<0.01),与T21呈显著正相关(P<0.05),与S22呈极显著正相关(P<0.01),说明κ-卡拉胶的添加使混合凝胶体系网络结构更加致密,不易流动水的比例相对提高,表现为宏观上的凝胶强度、硬度和咀嚼性的增大;另一方面,弹性与S21、T22和T2W呈极显著正相关(P<0.01),与S22呈极显著负相关,这与上文中弹性随κ-卡拉胶比例的变化相符,当体系中不易流动水所受束缚力较大时,胶体的弹性相对较弱。内聚性与T22和T2W呈极显著正相关性(P<0.01),说明不易流动水的流动性越强,凝胶体系的内聚性越大。黏性与T21呈极显著正相关性(P<0.01),说明结合水的流动性越强,凝胶体系的黏性越大,κ-卡拉胶通过结合水的流动性来影响混合凝胶的黏性。
2.5 动态黏弹性
储存模量G′是量度材料抵抗弹性形变力大小的尺度,损耗模量G″是材料发生黏性形变过程中损耗能量大小的量度,G′与G″一起反映材料黏弹性比例。不同κ-卡拉胶比例的混合胶液在降温与升温过程中G′与G″的变化如图8所示。
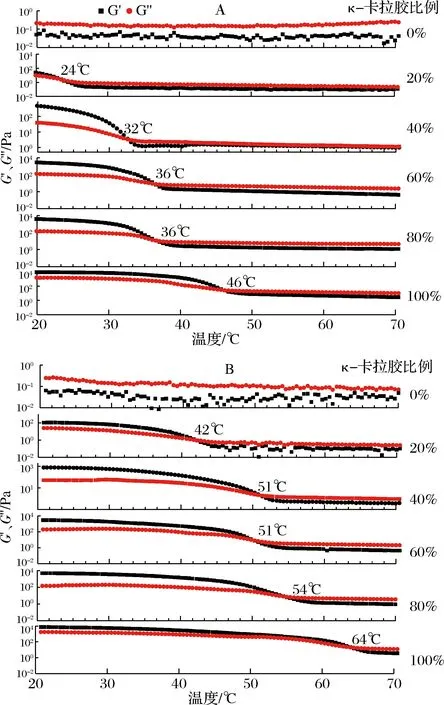
图8 部分混合凝胶液在降温(A)和升温(B)过程中G′和G″的变化Fig.8 Changes of G′ and G″ during partial cooling A and heating B of the mixed gel
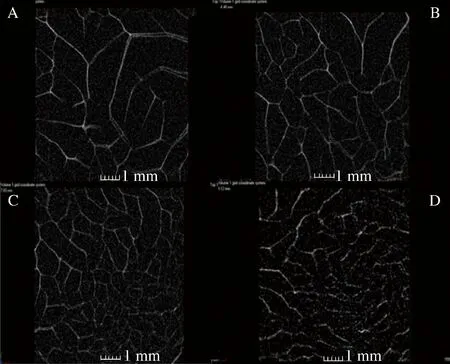
如图8-A所示,刚开始降温时,G′均小于G″,随着温度的降低,G′和G″均缓慢增大,当到达某一温度时,G′和G″迅速增加且G′增加幅度大于G″,最终使G′大于G″,这说明随着温度的降低,混合凝胶由无规则线团向螺旋转变,体系开始由溶液向凝胶转变,弹性开始占主导作用。在降温过程中,G′和G″的交汇点的温度为胶凝温度,从图8-A可以看出,未添加κ-卡拉胶时,在整个流变实验过程中体系(3%明胶)的G′ 升温过程中G′与G″的变化可以反映胶液熔化温度的改变。由图8-B可知,与降温过程相反,当温度由20 ℃开始升高时,G′大于G″,且G′和G″随温度的升高而逐渐减小,表明凝胶体的网络结构开始溃散,当超过某个温度,二者迅速下降,出现交点后G′则小于G″,说明发生了“凝胶-溶胶”的转变。G″和G′的交汇点的温度为熔化温度。总体而言,熔化温度均高于图8-A中的凝胶温度,且体系中卡拉胶比例会影响胶体的熔化温度。随κ-卡拉胶比例的增大,混合体系的熔化温度逐渐升高,当κ-卡拉胶比例由20%增加至80%时,凝胶的融化温度由42 ℃增加到54 ℃,这说明κ-卡拉胶可有效增加复配胶的凝胶特性,使凝胶在较高温度下仍保持一定的三维网络结构,这使混合凝胶体系成型后的温度范围相对扩大,有利于其成型加工。 利用冻干后的凝胶样品进行CT断层扫描,重构后选择截取代表性切面,如图9所示。 图9 部分混合凝胶体系的CT扫描代表性切面图Fig.9 Representative CT scanning sections of some mixed gel systems注:图A、B、C、D分别为κ-卡拉胶比例分别为0、20%、60%、80%的凝胶,图中标尺刻度为1 mm κ-卡拉胶比例对混合凝胶的微观结构有显著影响。单纯明胶凝胶体系(图9-A)网状结构较为疏松,空腔较大且大小不均匀。随着κ-卡拉胶比例的增加,混合凝胶的网络结构趋向于致密且均匀,这与孙哲浩[19]观察到的明胶与κ-卡拉胶共混凝胶体的微观结构相符。随体系中κ-卡拉胶比例的增加,凝胶体系逐渐从以明胶为主体的网络结构向κ-卡拉胶为主的凝胶网络转变,2种大分子长链间的交互作用加强,分子间的联结程度增加,凝胶网络结构更加致密且均匀,这种结构也极大地改善了凝胶强度及硬度、咀嚼性等宏观力学性能[20]。 本文重点研究了κ-卡拉胶的比例对明胶凝胶体系凝胶强度、质构特性、水分分布、动态黏弹性和微观结构的影响,结论表明,随κ-卡拉胶比例的增加,混合凝胶体系的凝胶强度、硬度和咀嚼性呈良好的二项式上升趋势(R2>0.94),而凝胶弹性显著减小,内聚性的变化较小,黏性和回复性先下降后上升,且在κ-卡拉胶比例为30%时达到最低值。在T2多组分弛豫图谱上,明胶、κ-卡拉胶及κ-卡拉胶-明胶混合凝胶体系均出现了2个弛豫峰。当体系中κ-卡拉胶比例≤50%时,随其比例的增加,弛豫图谱整体左移,T22、T2W相对减小,而当κ-卡拉胶比例>50%后,各弛豫时间均相对稳定。S21呈二项式下降趋势(R2=0.996),而S22呈二项式增加趋势(R2=0.997)。相关性分析表明,凝胶强度、硬度、咀嚼性与S21呈极显著负相关(P<0.01),与S22呈极显著正相关(P<0.01),与T21呈显著正相关(P<0.05);与S22呈极显著正相关(P<0.01);弹性与S21、T22和T2W呈极显著正相关(P<0.01),与S22呈极显著负相关;内聚性与T22和T2W呈极显著正相关性(P<0.01)。κ-卡拉胶比例对混合凝胶的动态黏弹性和微观结构有显著影响。单纯明胶凝胶体系无明显的胶凝化,网状结构较为疏松,随着κ-卡拉胶比例的增加,胶凝温度及熔化温度均相对升高,混合凝胶的网络结构趋于致密且均匀,这种结构也极大地改善了凝胶强度及硬度、咀嚼性等宏观力学性能。2.6 凝胶微观结构
3 结论
