高效液相色谱法测定卡马西平9个杂质方法的评价
2019-05-21陈运动
王 宇 陈运动 高 政
1.江苏天士力帝益药业有限公司,江苏淮安 223002;2.江苏慈星药业有限公司,江苏宿迁 223800
卡马西平(Carbamazepine)是抗癫痫的常用药物,是癫痫精神运动型发作和三叉神经痛的首选药物。在卡马西平的有关物质控制方面,中国药典与各国外药典均使用氰基色谱柱。在CP、USP与EP的色谱条件方法下,运用高效液相法检验时,卡马西平主峰与EP药典规定杂质A以及杂质B与杂质D之间的分离度达不到合格要求。参考文献[1-7]方法,现建立此有关物质的HPLC分析方法,对其验证,证明其方法专属性强,准确可靠。
1 仪器与试剂
1.1 仪器
AUW120D型电子天平(日本岛津公司),SQP型电子天平(赛多利斯公司),安捷伦1260型高效液相色谱仪,沃特世2695/2998型高效液相色谱仪。
1.2 试剂试药
甲醇色谱纯(美国 TEDIA 公司,17125121),乙腈色谱纯(美国 TEDIA 公司,18035024),甲酸分析纯(上海苏懿化学试剂有限公司,20170203),三乙胺分析纯(国药集团,20160323),杂质A对照品(Lot:016326.0,含 量:99.98%,European),杂质 B对照品(Lot:AJ-028-24,含 量:99.22%,Aocs),杂 质 C 对 照 品(Lot:ESC-037-26,含 量:99.91%,Aocs),杂质 D 对照品(Lot:1205821,含量:98.76%,REFERENCE),杂 质 E对 照品(Lot:AJC-058-24,含量:99.83%,Aocs),杂 质 F对 照品(Lot:AJC-058-25,Aocs),杂质 G 对照品(Lot:AJC-118-25,含量:99.08%,Aocs),酰氯亚氨基二苄(Lot:2018051301,含量:99.64%,江苏慈星药业有限公司),卡马西平(Lot:Y201801003,江苏慈星药业有限公司)。
2 实验方法与结果
2.1 色谱条件
流动相A:1000mL水中加三乙胺0.5mL甲酸0.5mL,混匀抽滤;流动相B:1000mL乙腈中加甲酸0.25mL,混匀,抽滤,按下述梯度条件测试。见表1。
检 测波 长:230nm,流速:1.0mL/min,进样量:20μL,柱温:30℃;色谱柱:月旭XS-C18(4.6mm×250mm,5μm)。

表1 流动相梯度洗脱明细表

表2 降解试验物料平衡考察结果
2.2 测定法
溶液配制。供试品溶液:取本品适量,精密称定,用甲醇-水(1∶1)定容,制成0.5mg/mL。
对照溶液:取供试品溶液2~50mL容量瓶,加甲醇-水(1∶1)溶解并稀释至刻度,摇匀,精密量取5mL,置50mL量瓶中,用甲醇-水(1∶1)稀释至刻度。
对照品储备液:取酰氯亚氨基二苄、杂质A、杂质B、杂质C、杂质D、杂质E、杂质F、杂质G对照品各20mg,精密称定,分别置100mL量瓶中,加甲醇50mL溶解后,用水稀释至刻度,分别制成每1mL中约含200μg的杂质储备溶液。
对照品定位溶液:分别精密移取杂质A、杂质E对照品储备液0.75mL,酰氯亚氨基二苄,、杂质B、杂质C、杂质D、杂质F、杂质G对照品储备液0.5mL,分别置100mL量瓶中用稀释剂定容。在分别移取5mL到10mL量瓶中用稀释剂定容(杂质A,杂质E为0.75μg/mL,酰氯亚氨基二苄、杂质B、杂质C、杂质D、杂质F、杂质G为0.5μg/mL)。
对照品混合溶液:分别精密移取杂质A、杂质E 对照品储备液0.75mL,酰氯亚氨基二苄、杂质B、杂质C、杂质D、杂质F、杂质G对照品储备液0.5mL,置同一100mL量瓶中用稀释剂定容。在移取5mL到10mL量瓶中用稀释剂定容。
系统适用性溶液:分别精密移取杂质A、杂质E对照品储备液0.75mL,酰氯亚氨基二苄、杂质B、杂质C、杂质D、杂质F、杂质G对照品储备液0.5mL,供试品储备液25mL,置同一100mL量瓶中用稀释剂定容。在移取5mL到10mL量瓶中用稀释剂定容。
精密量取系统适用性溶液、对照品混合溶液、供试品溶液、对照溶液各20μL,分别注入液相色谱仪,记录色谱图。系统适用性溶液中各杂质分离度要≥1.5,供试品溶液中如有与对照品溶液中杂质保留时间一致的色谱峰,按外标法以峰面积计算。
2.3 专属性
分别取本品50mg,精密称定,置50mL量瓶中,用溶剂甲醇-水(1∶1)稀释,在强碱、强酸、强氧化、高温条件下破坏后,室温放置24h;在光照条件下破坏5d进行强降解实验。同法制备相应破坏的空白溶液。精密量取各降解实验溶液20μL注入液相色谱仪,记录色谱图,报告溶剂对检测的干扰,报告系统适用性溶液中杂质的分离情况,计算物料平衡。试验结果表明溶剂不干扰其他杂质和主成份检测,对照品混合溶液中各杂质间分离度>1.5。强制降解试验中主要降解产物的峰纯度合格,物料平衡在90%~110%之间。见表2、图1。
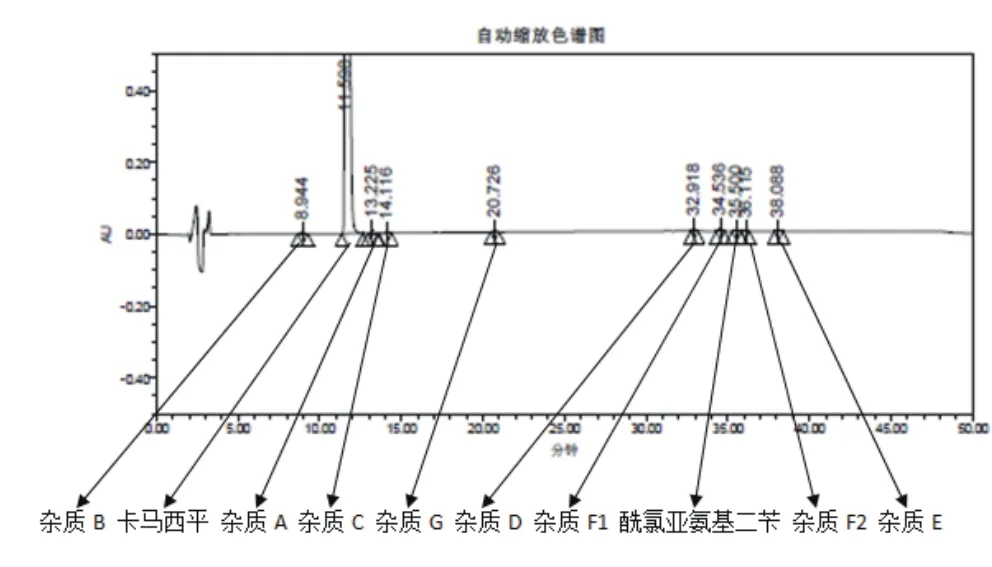
图1 分离度色谱图
2.4 检测限与定量限
精密量取检测限溶液和定量限溶液各20μL注入液相色谱仪,记录色谱图。见表3。
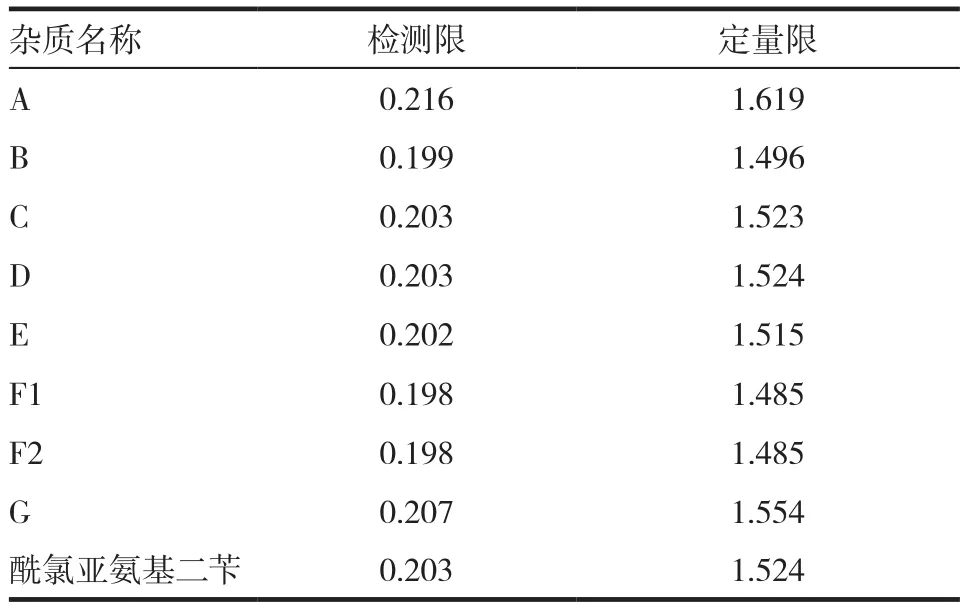
表3 检测限、定量限结果(μg/mL)
2.5 线性范围
配制每1mL中各约含10μg的对照品储备液。取对照品储备溶液配制各杂质项下线性溶液,各精密量取20μL,注入液相色谱仪。记录各浓度的色谱峰面积,以峰面积A对浓度C(μg/mL)进行线性回归,得出各杂质的线性回归方程,线性范围。杂质A为0.5395~3.2370μg/mL(y=20.012x+0.794,r=0.9962,n=6),杂质 B 为 0.4985~ 2.9910μg/mL(y=60.679x+1.2167,r=0.9995,n=6),杂 质 C 为0.5075~ 3.0450μg/mL(y=67.349x-0.8307,r=0.9995,n=6),杂质 D 为 0.5080~ 3.0480μg/mL(y=89.659x-4.208,r=0.9999,n=6),杂 质 E为 0.5050~ 3.0300μg/mL(y=32.231x+3.8737,r=0.9973,n=6),杂质 F1为 0.4950~ 2.9700μg/mL(y=62.706x+0.7887,r=0.9998,n=6),杂 质 F2 为0.4950~ 2.9700μg/mL(y=14.772x+3.0857,r=0.9962,n=6),杂质 G 为 0.5180~ 3.1080μg/mL(y=61.868x+6.2873,r=0.9986,n=6), 杂质酰氯亚氨基二苄为0.5080~3.0480μg/mL(y=32.855x+1.1273,r=0.9986,n=6),各杂质线性方程的相关系数R2≥0.990,y轴截距在100%响应值的25%以内,线性良好。
2.6 准确度
溶液配制:
对照品储备液1:用甲醇-水(1∶1)稀释,制成每1mL中含杂质A和杂质E各约10μg的对照品储备液。
对照品储备液2:用甲醇-水(1∶1)稀释,制成每1mL中各约含10μg的酰氯亚氨基二苄、杂质B、杂质C、杂质D、杂质F和杂质G对照品储备液。
样品溶液:取本品50mg,精密称定,置50mL量瓶中,精密称定,加甲醇25mL使溶解,用水稀释至刻度,摇匀,制成每1mL中约含卡马西平1mg的供试品溶液。
准确度溶液:取本品50mg,精密称定,置50mL量瓶中,按限度水平50%、100%、150%的相应浓度,精密量取杂质对照品储备液1和杂质对照品储备液2,分别加入各已有样品的量瓶中,加甲醇25mL使溶解,用水稀释至刻度,摇匀,滤过,即得。每浓度水平的准确度溶液平行配制3份。
测定:精密量取上述溶液各20μL注入液相色谱仪,记录色谱图。分别计算各个浓度水平的单个和平均回收率和相对标准偏差,各浓度3份溶液中单个和平均回收率在90%~110%之间;相对标准偏差(RSD)≤10%。
2.7 精密度
重复性试验取准确度项下溶液,参考文献[8-11]方法,重复进样6次,各杂质对标准偏差(RSD)均≤8%。中间精密度试验为不同研究员,在不同日期,使用不同液相色谱仪,按重复性试验同法操作,各杂质相对标准偏差(RSD)均≤10%。
2.8 耐用性
取杂质储备液、对照品混合溶液和供试品溶液,对照溶液,于室温条件和冷藏条件下放置,参考文献[12-15]方法,在一定的时间间隔取样测定,计算杂质储备液、对照品混合溶液在不同稳定性时间点各峰面积,计算待测物质峰面积RSD,比较供试品溶液在各稳定性时间点的杂质检出情况。
改变色谱条件:与正常条件相比较,在改变色谱柱温度为 25℃、35℃、流速 0.9mL/min、流速1.1mL/min以及更换色谱柱条件下,考察供试品溶液中杂质的变化。对照品储备液、对照品溶液稳定性在相应稳定性时间点峰面积相对标准偏差(RSD)均≤5%,杂质<0.2%,绝对差值在±0.05%,杂质检出个数没有差异。
3 讨论
通过对此卡马西平有关物质方法的验证,各项验证数据均符合中国药典标准规定,从而证明此方法在准确度、专属性、耐用性等方面表现良好。目前,各个国家药典中均选用氰基色谱柱,当氰基柱在使用四氢呋喃做流动相时,色谱柱的寿命很短,且长期使用柱效下降。其中,USP方法为超高效液相法,对仪器仪器与耗材要求严格,成本较高。而运用CP方法与EP方法高效液相检验时,卡马西平主峰与杂质A以及杂质B与杂质D之间的分离度达不到合格要求,不能实现所有杂质的分离,导致杂质限度检验不准确。此有关物质的HPLC检查方法,方法专属性强,准确可靠,建议中国药典采用此方法进行卡马西平有关物质检查。
