NO在金属Ben(n=2-12)团簇表面的平行吸附
2019-04-29李淑萍王继刚
李淑萍, 孟 江, 王继刚
(1. 西藏民族大学信息工程学院, 咸阳 712082; 2. 东南大学材料科学与工程学院,南京 410083)
1 引 言
汽车尾气及工业废气对环境污染的一个重要来源就是 NO排放对空气的污染. 工业上一般采用含贵金属的催化剂以减轻污染物的排放[1,2]. 因此,研究NO在过渡金属,尤其是贵金属表面的吸附行为具有重要的实际意义[3-10]. 同时,由于过渡金属团簇表面存在大量的悬挂键,在吸附NO等小分子时表现出了更好的吸附强度,是人们在理论上关注的重点课题[7, 11].
但是不论块体还是团簇,过渡金属,特别是某些贵金属(Pt, Pd等),由于价格昂贵限制了其应用规模. 因此能否寻找合适的简单金属,探讨其吸附NO等小分子时的相关性质,更具现实意义.
我们知道,二价碱土金属铍(Be)是原子反应堆中快速中子的主要来源,研究氢与铍的相互作用不仅是核聚变反应的重要课题,而且在其它领域,如铍在氢燃料电池制备等方面,也有重要的应用[12, 13]. 因此,块体Be与H2、H2O等小分子的相互作用近年来已经成为一个热点领域[14-17]. 我们发现,以上所有报道,关注的都是块体Be (0001) 面的吸附小分子的行为,然而结果表明,其吸附H2、H2O时,吸附强度是很弱的.
Be 在小尺寸团簇范围内,不仅具有复杂的表面,而且原子间相互作用较弱,类似范德瓦尔斯作用力,这样吸附NO等小分子时,与其它过渡金属团簇相比,吸附位置、强度及相应的电子性质有何差异,是我们感兴趣的课题.
小分子不论吸附在块体还是团簇表面,最重要的是确定相应体系的能量最低结构. 以NO吸附为例,首先要计算N端,O端还是N,O同时吸附在不同的位置上的能量,以此确定体系的基态结构. 这是研究其它电子性质的基础. 在所有研究过渡金属块体(Rh, Pt, Ir等)吸附NO的文献中[3-10],给出的基态结构均为N端吸附于面位,桥位或者空位而形成的. 受限于实验条件,团簇吸附NO的研究主要是第一性原理计算. 同样,Rh,Y等团簇吸附NO的最低能量结构仍然是N端吸附于桥位的构型[7, 11]. 其它吸附位,相关文献未做任何报道.
在研究团簇吸附小分子时,由于其表面复杂,需要考虑的初始结构很多. 以NO为例,N端吸附,O端吸附以及线型的NO分子平行吸附于团簇某个表面时的概率是完全相等的. 只有考虑到以上所有初始构型,才能最终确定最低能量结构.
我们在n=2-12的尺寸范围内,充分考虑各种吸附位置的情况,通过结构优化,发现NO在Ben团簇某个面平行吸附时,相应体系的能量最低,而N端吸附Be-Be桥位的结构,其能量要明显高于平行吸附. 这与以往通过理论计算报道的NO在过渡金属(或团簇)表面的吸附行为结果是不同的. 希望我们的计算结果能为相关研究提供有意义的借鉴.
2 计算方法
采用密度泛函理论(DFT)下的广义梯度近似 (GGA),用DMOL3软件包[18]对全部结构进行构型优化和电子性质计算. 在GGA方案中,选择BLYP方法[19]和全电子基组进行计算[20]. 在计算过程中,电子结构计算以体系的能量是否收敛为判据,精度优于10-5a.u.. 结构优化以梯度,位移和能量是否收敛为判据,梯度和位移均优于10-3a.u.,能量收敛精度优于10-5a.u..
为了验证所用方法的有效性,我们首先计算了二聚体Be2的键长(2.468 Å),结合能(0.17 eV)与实验值(2.47 Å、0.11 eV)[21, 22]基本吻合. 对于优化后的自由NO分子,计算得到的键长为1.169 Å,与实验值1.151 Å[23]十分接近. 我们给出的频率(1842.11 cm-1)也与实验结果(1876 cm-1)[24]吻合, 说明所用方法对该体系是合适的.
3 结果与讨论
3.1 基态结构
为了确定BenNO (n=2-12)团簇的的基态构型,我们首先用同样方法计算得到了Ben(n=2-13) 团簇的基态结构,与其它理论计算相比,结果基本一致[25]. 然后,分两步设置NO吸附的初始结构. 其一,将NO分子平行吸附于于Ben团簇的某个面(对于对称性低的团簇,要考虑到所有面位). 其二,以NO的N端或者O端分别在Ben的端位、桥位、面位吸附NO. 对所有初始构型构型在GGA方案下进行几何优化和频率分析,对于有虚频的结果,通过结构的适当调整进行再优化,以确保得到的是稳定结构. 图1,给出了我们计算的Ben(n=2-13) 和BenNO (n=2-12) 团簇的基态构型.
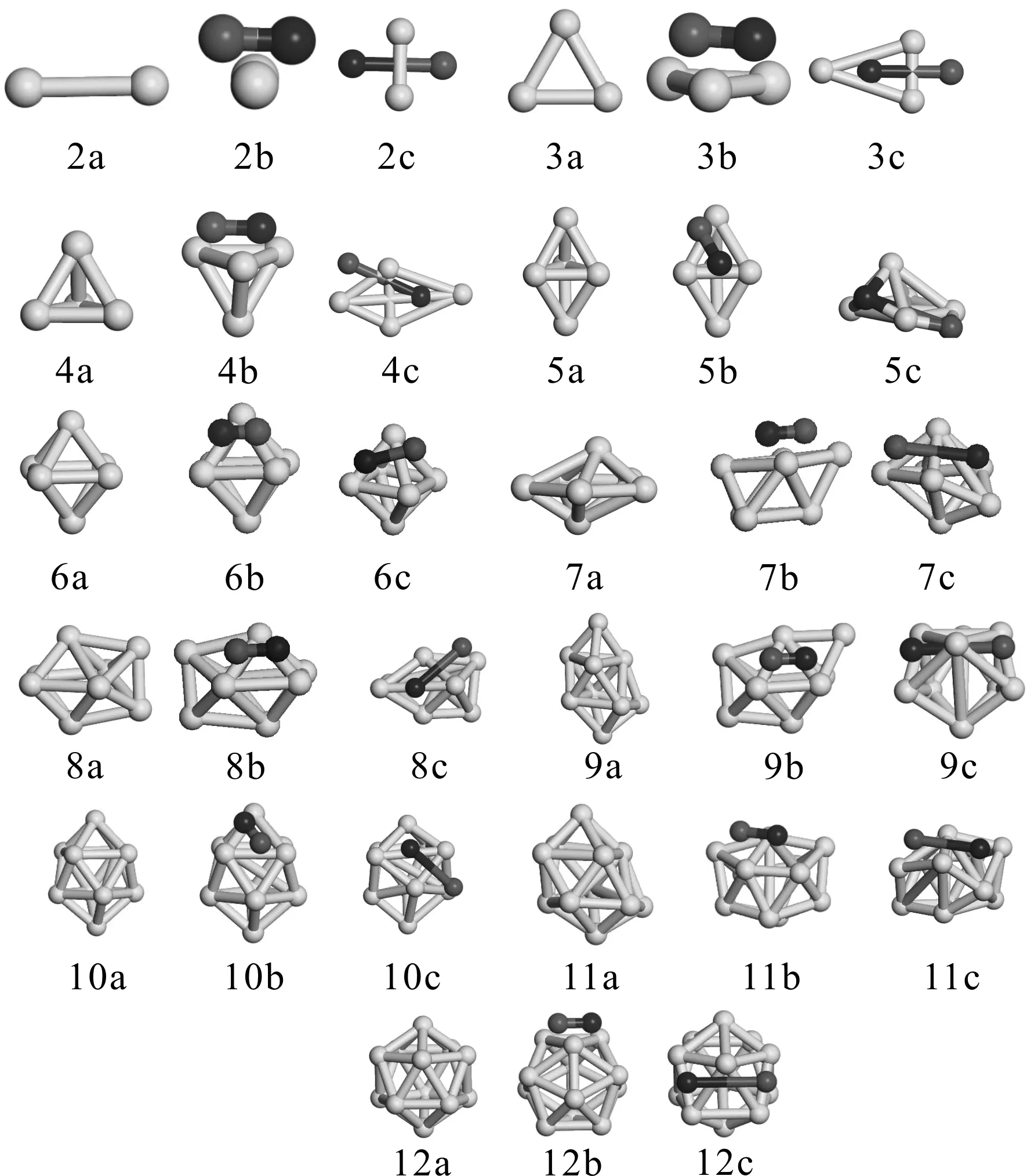
图1 Ben (n=2-13)和BenNO(n=2-13)团簇的基态结构 (na表示Ben基态结构,nb表示BenNO吸附NO时的初始构型,nc表示BenNO驰豫后的构型)Fig. 1 The geometry structures of Ben(n=2-13) and BenNO(n=2-13) clusters(na represent the ground state structures of Ben. nb and nc represent the initial structure and structure after relaxation, respectively.)
Be2吸附NO的初始构型,我们分别考虑了NO在空间垂直,平行,以及N端,O端吸附Be-Be桥位吸附的情况,驰豫后终态结果如图2c所示. 可以看出,Be2NO的基态构型为N,O原子对称地吸附于Be2桥位的构型,与自由的NO分子相比,N-O键长的伸长量达到了120%. 不过,吸附后二聚体Be2的键长变为1.805 Å,与吸附前相比(2.468 Å),缩短了约27%. 而N端吸附Be2桥位的构型与基态相比,能量高出了4.18 eV. 此时二聚体Be2的键长为1.944 Å,与吸附前相比同样缩短了.
n=3, 4, 5时,虽然尺寸不同得到的稳定构型数目不一样. 但是,BenNO(n=3, 4, 5)的基态都是初始构型设置为NO平行吸附于Ben的某一表面(由对称性可知,这三个尺寸只有一个吸附面位)时经结构驰豫得到的. 很显然,吸附时,N,O原子倾向于同时与近邻Be原子成键,由此均导致了三个尺寸的主团簇结构发生彻底改变. 此时,N,O原子间距与自由的NO分子相比,显著伸长,特别是n=4时,伸长量超过了130%.
Be6为高对称性Oh的四角双锥构型. 由对称性可知,NO吸附于面位的初始结构只有一种, 驰豫后,主团簇结构并未改变,但是NO的N,O原子间距变大(约3.23 Å),O原子倾向于在Be-Be桥位成键. 计算表明,此为能量最低结构.
我们的计算给出的Be7的基态结构为对称性较低的五角双锥构型,这与文献[24]报道的结果是一致的. 然而,基态的Be7吸附NO时,不论怎样设置初始的吸附位置,结构驰豫后,要么收敛失败,要么计算的结构存在虚频. 即NO在基态Be7表面吸附时,没有稳定结构. 同样我们也得到了Be7的一个亚稳态构型(在Be6基态结构的表面带帽一个Be原子得到的结构),当NO平行吸附于四个Be原子的平行四边形表面时, 优化后得到了Be7NO的基态结构. 此时NO间距增大,N, O原子倾向于分别与近邻的三个Be原子表面成键.
从n=8开始,吸附NO时需要考虑的初始构型明显增多,计算表明,同一尺寸的不同面位,平行吸附时,驰豫后得到的结构能量差别不是特别大,多数都小于1 eV. 但是N端位吸附于面位或桥位时,所得结构,能量明显高于基态,差值在4 eV以上. 同时NO吸附均导致了主团簇基态构型的变化,特别是n=9, 11, 12时,结构畸变明显,团簇的对称性明显降低. 另外与其它尺寸相似,吸附均导致了N,O间距不同程度的伸长.
以上分析可以看出,当初始结构设置为NO分子平行吸附于Ben的某个面位时(n=2除外),经结构优化得到的构型为相应尺寸的能量最低结构. 几乎所有的尺寸,NO吸附导致了主团簇Ben结构发生畸变. 同时NO分子的N, O间距均有不同程度的伸长. 而N端吸附于Be-Be桥位的初始构型,驰豫后只是一个亚稳态. 这与Rhn, Yn吸附NO的情况是截然不同的[ 7, 11 ].
3.2 吸附强度和电子性质
研究团簇吸附小分子的吸附效率,必须考虑吸附能的变化,它可以反映出Ben团簇与NO分子之间的相互作用,吸附能的值越大, 说明NO分子更易于吸附于Ben表面,吸附效率越高. 其计算公式如下:
Eads=-[E(BenNO)-E(Ben)-E(NO)]
(1)

图2 BenNO (n=2-12) 基态团簇的吸附能Fig. 2 Adsorption energy of ground state structures of BenNO (n=2-12) clusters
图2给出了NO吸附于Ben团簇表面的吸附能随尺寸的变化. 可以看出,Ben团簇表现出了很好的吸附NO分子的能力,吸附能的值多数在9 eV附近震荡. 即使吸附能最小的尺寸(n=2, 6)其值也达到了6 eV. 与Yn团簇(其吸附能分别在6 eV,9 eV两个值附近震荡)相比,简单金属Be表现出了更好的吸附NO的能力. 结合图2所示结构,我们发现,n=6时,NO吸附对主团簇结构的影响也是最小的. 很显然,与其它尺寸相比,NO的N原子虽然同时吸附于三个Be原子面位,但是O原子则吸附于Be-Be桥位,这样不仅主团簇结构变化不大,而且对N-O间距的影响也是有限的.
分析键长变化是团簇吸附NO小分子时,讨论主团簇催化能力的重要步骤. 图4给出了BenNO团簇中NO的N-O键长随尺寸增大的变化规律. 可以看出,与驰豫后自由的NO分子的N-O键长(1.169 Å)相比,吸附后,除n=6以外,N-O键长均出现了明显的伸长.n=3, 8, 10, 11时,长度约为2.6 Å.n=4, 5, 9, 12时,长度约2.9 Å.与1.169 Å相比,伸长量分别达到了122%和148%. 这种变化规律与Yn吸附NO时情况不同. NO在Y3, Y5, Y8团簇表面吸附时,N-O间距伸长量达到了300%,而其它尺寸却只有20%[11]. 可见,简单金属在团簇尺寸内吸附NO分子的行为与Y等过渡金属是不同的.
这种吸附行为应该与Be团簇的性质以及N, O原子的轨道杂化有关. 如前文所述,Be原子在小尺寸范围内,原子间相互作用很弱. 吸附NO后,N-Be, N-O成键概率明显要高于Be-Be间作用力. 此时,N, O原子以sp3杂化方式分别形成四个杂化轨道,其中N原子的三个单电子轨道与近邻的三个Be原子成键后,还有一对孤对电子不参与成键. 而O原子四个杂化轨道中两个单电子轨道与近邻的Be原子成键后,还有两对孤对电子不参与成键. 根据价电子对互斥理论,孤对电子与孤对电子间存在最大的排斥力[26],由此不仅增强了N-Be, N-O的成键能力,而且导致了N、O原子间距离急剧增大.
能隙(Gap)是反应团簇化学性质的重要物理量. 它是团簇最高占据分子轨道 (HUMO) 和最低未占据分子轨道(LUMO) 的能级之差,反映了电子从HUMO向LUMO发生跃迁能力大小. 一般来说,能隙越小,相应的团簇化学活性更好. 我们在图5计算给出了BenNO团簇的能隙随尺寸的变化. 同时为了比较吸附对主团簇化学活性的影响,我们在 图中也给出了相应的中性Ben团簇的能隙值. Be原子为闭壳层结构,小尺寸的Be团簇具有较好的化学稳定性. 我们给出的Ben(n=2-12)团簇的能隙值,与已有的研究结果[23]相比,两者在所用软件以及计算方法不同的情况下,整体变化趋势是完全一致的. 可以看出,Be4, Be10团簇,其值为局域最大(均超过了1 eV), 这与其幻数行为完全吻合,也与已有的研究结果是一致的[23]. 吸附NO后,BenNO团簇的能隙与吸附前相比,可以分为两种情况.n=2, 3, 12时,吸附导致了主团簇能隙值减小. 减小的幅度均超过了0.5 eV. 其它尺寸,两者的变化趋势则几乎完全相似.n=4,7,10时,相应的BenNO团簇均为局域最大,特别是Be4NO的能隙值超过了2 eV,具有很好的化学稳定性. 同时,我们也注意到,Be6NO的能隙值在所研究的尺寸内是最小的,约为0.024 eV,虽然主团簇Be6也为局域最小,其值为0.24 eV,但是吸附NO后,能隙值减小了近10倍,表现出了较好的化学活性.
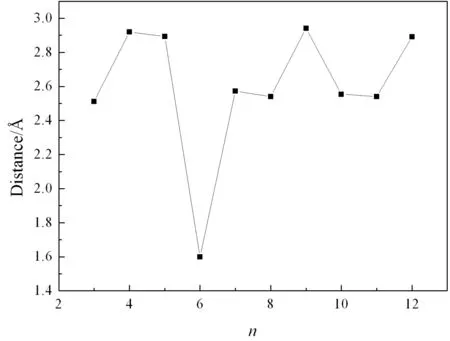
图3 BenNO(n=2-12)基态团簇中N, O的原子间距Fig. 3 Distance between N and O atoms of ground state structures of BenNO(n=2-12) clusters
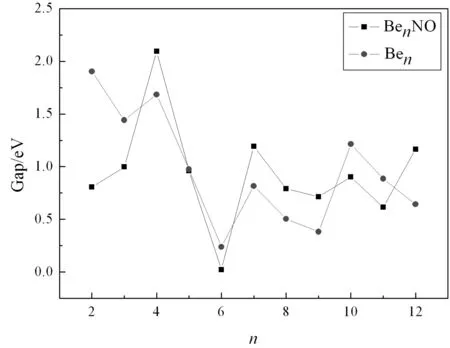
图4 Ben,BenNO(n=2-12)基态团簇的能隙Fig. 4 Energy gap of ground state structures of Ben and BenNO(n=2-12)
3.3 成键特性及电荷转移
为了直观地观察原子间相互作用,我们在图5给出了n=4, 6, 7, 12时相应团簇的差分电荷密度分布图. 可以看出不同的尺寸,吸附NO以后,N,O原子与Be原子间作用存在差异. Be6NO团簇的电子倾向于集中分布于N-Be, O-Be之间,Be-Be间电子出现的概率很小. 其它尺寸不仅N,O原子与相应团簇间有密集的电子分布,而且Be-Be间也可观察到明显的电子. 很显然,相比于其它尺寸,n=6时相应团簇的稳定性相对要弱一点. 这与前文吸附能的表述是一致的.
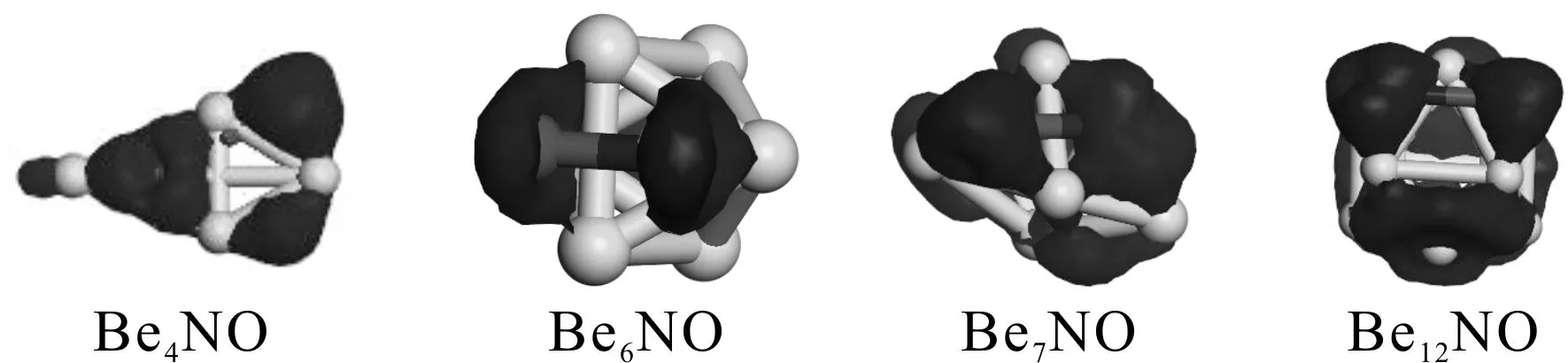
图5 Be4NO、Be6NO, Be7NO、Be12NO基态团簇的差分电荷密度Fig. 5 Deformation charge density of ground state structure of Be4NO、Be6NO, Be7NO、Be12NO clusters
为了进一步理解NO与Ben团簇相互作用的物理实质,需要研究不同尺寸的团簇态密度的分布情况. 我们以n=4, 6为例在图8给出了团簇的总态密度(DOS)分态密度(PDOS).
由总态密度图可以看出,在费米能级附近(-0.2-0.2 eV),Be4NO和Be6NO都分布有若干个尖锐的峰,电子表现出了很强的局域性. 相比之下,n=4时,杂化峰更尖锐,特别是在费米能级以下约-0.15 eV处,Be4NO存在一个杂化峰,约450 eV-1,与n=6相比,其值高出了约100 eV-1. 另外,两者均可观察到明显的贋能隙. 相比之下,Be4NO的赝能隙更宽. 一般来说赝能隙越宽,原子间成键能力越强,相应的体系具有更好地稳定性. 因此,Be4NO比Be6NO具有更好的稳定性,这与前文吸附能,能隙给出的计算结果吻合.
分态密度可以反应不同轨道对成键的影响. 可以看出,在费米能级附近-0.2 eV—0.2 eV范围内,不同的轨道对成键的贡献是不一样的. Be4NO和Be6NO的杂化峰中,p轨道对轨道杂化都起主导作用. 但是,进一步分析可以看出,在-0.25-2.5 eV能级处,Be6NO在成键过程中,d轨道成分明显增多了. 我们知道,对于Be, N, O原子来说,电子被激发到d轨道时,相应电子具有很好的化学活性,其上电子越多,相应的Be6NO具有更好的化学活性.

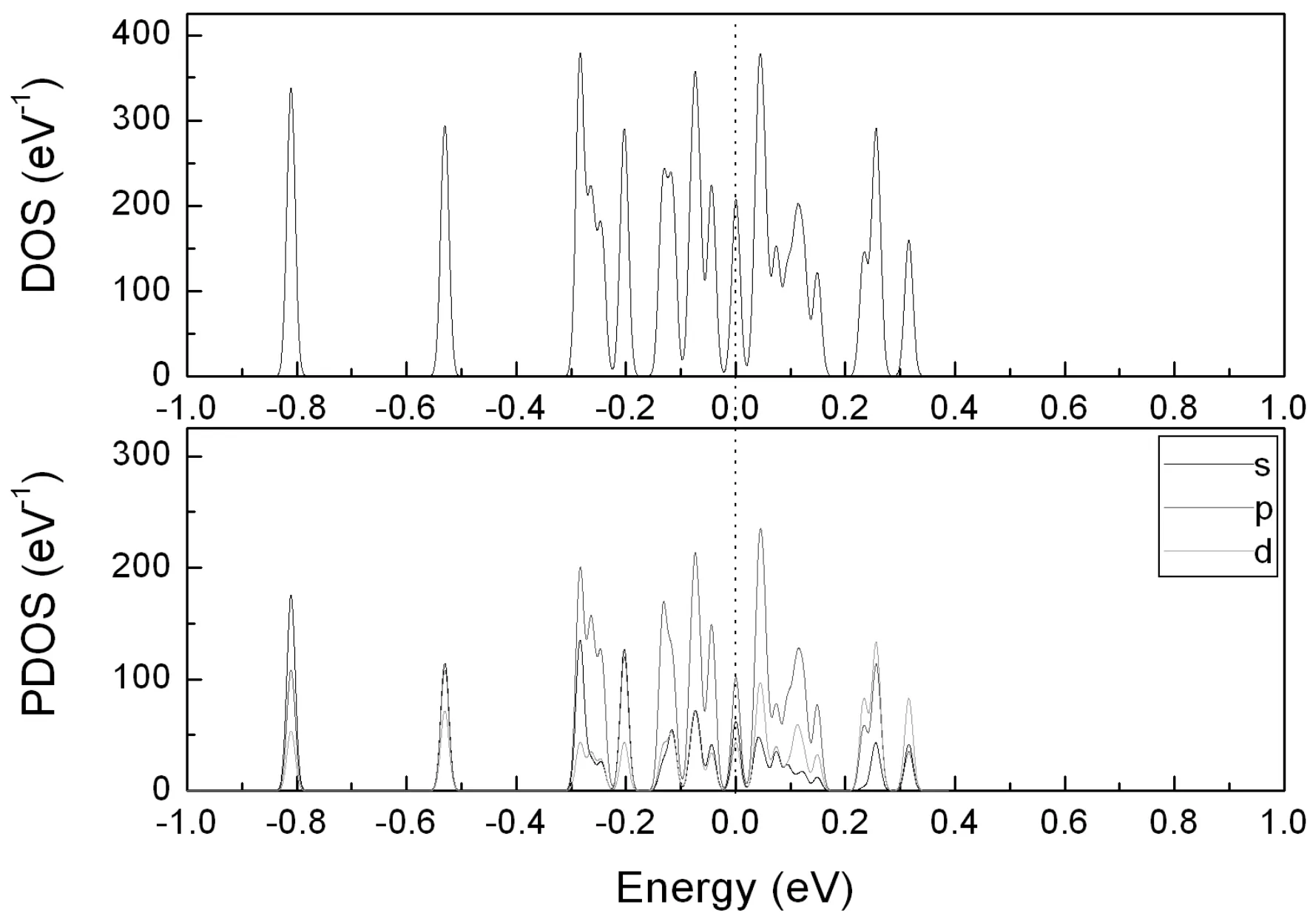
图6 Be4NO, Be6NO基态团簇的总态密度和分态密度Fig. 6 Total state density and partial state density of ground state structures of Be4NO, Be6NO clusters
4 结 论
运用密度泛函理论,研究了BenNO (n=1-12)团簇的稳定结构和电子性质. 结果表明:当NO分子平行吸附于Ben(n=1-12)团簇得某一表面时,经结构驰豫得到了相应团簇的基态结构. 在多数情况下,NO平行吸附使主团簇Ben结构发生明显畸变. 此时,Ben团簇表现出了很好的吸附NO分子的能力,吸附能的值至少在6 eV以上. 吸附后NO分子的N-O间距显著伸长,伸长量均超过了100%,为典型的解离性吸附. 成键特性分析表明,多数尺寸下,N,O原子倾向于同时于近邻Be原子成键,此时N, O键断裂,相应的团簇表现出了很好的稳定性.
