HSF1基因缺失通过上调microRNA-195a-3p加速压力超负荷下心脏重构*
2019-03-21王时俊杨继娥马雷雷崔兆强邹云增葛均波
王时俊,徐 磊,赵 刚,杨继娥,马雷雷,崔兆强,邹云增,葛均波
(复旦大学附属中山医院, 上海市心血管病研究所, 上海 200032)
热休克反应(heat shock response)是普遍存在机体内的一种分子应激性防御反应,一般通过激活细胞核内热休克转录因子1(heat shock transcriptional factor 1,HSF1),继而转录合成一组热休克蛋白(heat shock proteins,HSPs),HSPs 具有分子伴侣功能,可参与细胞内多种蛋白质的折叠、聚合、转运和信号传递等生理作用[1-2]。在我们既往的研究中发现,HSF1过表达可以显著减少心肌细胞凋亡、纤维化和心肌肥厚,而小鼠HSF1基因缺失则会加重压力超负荷下的心脏重构[3]。同时,HSF1缺失导致的心肌重构并不因HSP70和HSP27等热休克蛋白过表达而发生逆转,提示HSF1可能通过HSPs非依赖的调控机制,促进心肌代谢,细胞存活和改善心脏重构[4]。我们进一步的研究证实,HSF1缺失主要导致了心脏的血管新生功能障碍,从而引起机械应力下的心肌细胞缺血缺氧,加速了细胞肥大反应[3, 5]。然而,HSF1缺失如何导致血管新生障碍,其背后的病理分子机制还不甚清楚。
因此,在本研究中我们拟通过HSF1调控的表观遗传学改变,筛查HSF1敲除前后的微小RNA(microRNA, miR)表达芯片谱差异,试图找到在HSF1基因敲除小鼠的心肌组织中明显上调的microRNA分子,并探究其与心脏血管新生功能障碍之间的联系,阐明HSF1基因缺失加重压力超负荷下心脏重构的疾病分子机制,并明确可干预的分子靶点。
材 料 和 方 法
1 实验动物
SPF级的HSF1基因敲除(knockout, KO;HSF1-/-)小鼠和C57BL/6背景野生型(wild-type, WT)对照小鼠,雄性,8~10周,20~25 g,饲养于复旦大学附属中山医院实验动物中心,动物许可证号为:SYXK(沪)2016-0006。本实验所涉及的动物实验相关操作和动物护理均依据美国国家卫生研究院发表的实验动物保护和使用准则(NIH指南发布:85-23号出版物,1996修订)以及复旦大学实验动物伦理委员会的相关规定。
2 主要方法
2.1小鼠压力超负荷模型的构建 通过主动脉弓缩窄(transverse aortic constriction,TAC)方法实现小鼠心脏的压力超负荷。选取8~10 周龄的雄性HSF1-/-小鼠(并以同龄C57BL/6野生型小鼠作对照),腹腔注射氯胺酮(100 mg/kg)和甲苯噻嗪(5 mg/kg)麻醉小鼠,行气管插管,并接入呼吸机(使呼吸频率保持在110 min-1,潮气量0.4~0.5 mL),小鼠左侧胸部褪毛后取仰卧位固定于操作板,乙醇消毒后逐层分离皮肤和肌肉,沿胸骨上窝至左第2肋水平撑开5 mm并固定撑开位,分离胸腺暴露主动脉弓,将升主动脉开口上3 mm处血管与27号垫针一起结扎,30 s后拔出垫针,观察到主动脉近心端的波动增强,逐层缝合并消毒。假手术只开胸不做主动脉弓结扎。
2.2小鼠心脏超声心动图检查 TAC术后4周行小鼠心脏超声检查。小鼠左侧胸部褪毛,麻醉后仰卧位固定在加热恒温的超声检查台上,并接入呼吸机。采用30 MHz的高频探头(Vevo 770,VisualSo-nics),将M型光标的垂直线置于室间隔和左心室后壁乳头肌水平,输出M型超声图像记录和心脏功能相关指标。
2.3心肌组织免疫组化染色 TAC术后4周,小鼠麻醉处死后心脏取材,用4%多聚甲醛固定,经脱水、透明、石蜡包埋后制成切片,切片行HE染色和抗CD31抗体染色(ABC法)[6]。HE染色切片进一步用Leica图像软件计算细胞横截面积(cross-sectional area, CSA)并作定量分析。
2.4心肌微血管内皮细胞血管生成实验 取8~10周龄成年C57BL/6小鼠经5%异氟醚麻醉后脱颈处死,分离左心室组织并剪切1 mm×1 mm×1 mm组织块,滴加1 mL胎牛血清,均匀接种于培养皿(预先在皿底铺鼠尾胶,超净台中风干过夜),组织块间隔2 mm,37 ℃含5% CO2培养箱静置4 h后,追加20%高糖DMEM(改良Eagle培养基),再培养18~24 h后组织块周围有细胞爬出,即原代微血管内皮细胞,利用抗CD31抗体染色鉴定内皮细胞,48~72 h后大量细胞爬出,去除组织块后继续培养36 h后待细胞生长铺满培养皿,用0.125%胰酶(含0.02%EDTA)消化细胞并传代,进行细胞成管实验[7]。
Matrigel(No.356234,BD Biosciences)铺24孔板底,超净台中风干过夜,第2天在板上接种微血管内皮细胞,1%胎牛血清含高糖DMEM,37 ℃、 5% CO2培养箱中孵育18 h后,在显微镜下观察管腔样结构。计数5个随机视野的管腔数量,并作统计分析。
2.5PRKAA2基因3’-UTR功能预测和评价 首先通过TargetScan6.2生物信息软件预测miR-195a-3p的靶基因,结合血管新生信号通路进行筛选;从NCBI数据库获取选定靶基因的小鼠编码序列,结合软件分析其3’-UTR区域与microRNA互补的序列片段,分别设计野生和突变(无义序列)3’-UTR核酸片段,通过亚克隆将2种3’-UTR连接到pMIR-REPORTTM系统,采用Luciferase报告系统对预测位点的实际结合能力进行测定。具体步骤为:微血管内皮细胞经无血清预处理后接种于24孔板,通过siPORTTMXP-1转染试剂(AM4507, Applied Biosystems)将上述构建的pMIR-WT-3’-UTR和pMIR-Mut-3’-UTR载体(对照质粒)分别转染微血管内皮细胞,转染浓度为100 nmol/L;然后用miR-195a-3p模拟物(mimic)刺激细胞,细胞继续培养48 h后,采用双萤光素酶报告基因系统(Promega)检测荧光强度。将各组的海肾萤光素酶与萤火虫萤光素酶的相对比值(Rluc/Luc)与对照孔的Rluc/Luc(校正为1)进行统计分析,判断预测的3’-UTR是否为miR-195a-3p的结合位点。
2.6RT-PCR和real-time PCR实验 将左心室心肌组织反复剪碎后,溶解于1 mL TRIzol(Invitrogen)溶液中并匀浆,然后加入200 μL氯仿,上下混匀30 s后室温静置10 min后11 000 r/min离心15 min,取上清加入500 μL的异丙醇,上下轻轻混匀数次,将样本置于-20 ℃。30 min后9 000 r/min离心10 min,弃除上清后将EP管中沉淀置于60 ℃烘干箱2 min,加入20 μL 1×TE缓冲液溶解总RNA。然后,按照PrimeScriptTMRT试剂盒(TaKaRa)说明书进行RNA逆转录,再按照SYBR® Premix Ex TaqTMII(TaKaRa)进行real-time PCR或普通PCR扩增,引物序列见表1。MicroRNA表达水平用2-ΔΔCt法计算;心肌肥厚标志物心房钠尿肽(atrial natriuretic peptide, ANP)和β-肌球蛋白重链(β-myosin heavy chain, β-MHC)的PCR产物用琼脂糖凝胶电泳进行半定量分析。
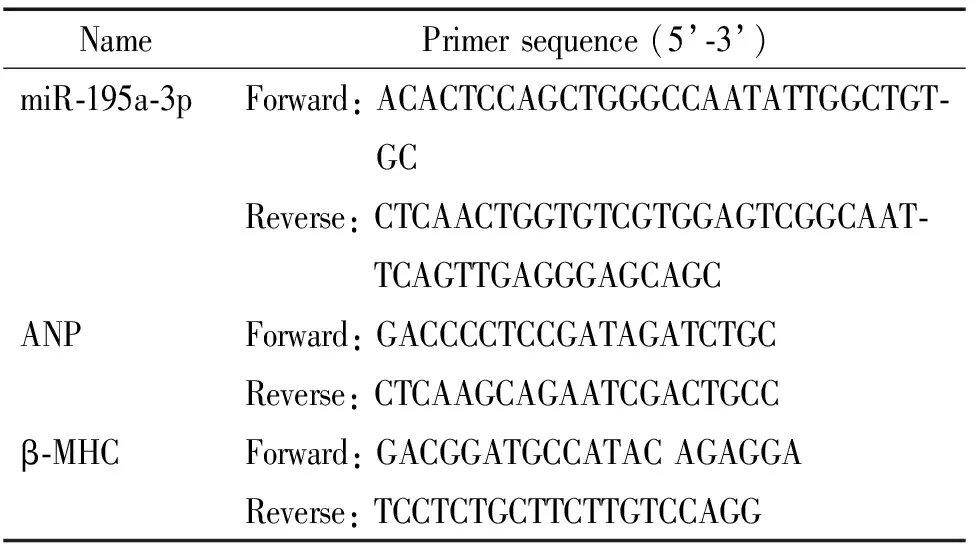
表1 PCR引物序列
2.7Western blot实验 选取左心室心肌组织,剪碎后匀浆并裂解于1 mL预制的RIPA蛋白裂解液中(含50 μL PMSF,蛋白酶抑制剂)。室温静置20 min后,采用12 000 r/min高速离心于4 ℃离心 15 min,取上清液每个蛋白样本加入含溴酚蓝的6×电泳缓冲液10 μL,95 ℃水浴3 min。取变性后的蛋白样品30 μL上样,在10% SDS-PAGE分离2 h,参考蛋白marker的电泳位置截取分离凝胶,将蛋白转印至PVDF膜(Millipore)。随后,PVDF膜用5%牛血清白蛋白(BSA)封闭1 h,加入 I 抗(1∶2 000)4 ℃孵育过夜;用1×PBST洗涤PVDF膜3次后加入辣根过氧化物酶(HRP)标记的II 抗(1∶500)室温孵育2 h;再用1×PBST洗涤PVDF膜3次后加入高敏ECL显影液;2 min后在ChemiDocTM化学发光成像仪(Bio-Rad)曝光和定量分析免疫条带。
2.8MicroRNA芯片筛查 采用华联生物提供的microRNA表达芯片谱筛选HSF1基因缺失对小鼠心脏microRNA的影响。分别取HSF1-/-小鼠和野生型小鼠左心室组织,利用TRIzol提取总mRNA(如2.5所述);通过microRNA表达谱芯片筛查,筛选条件为HSF1-/-较野生型对照表达阈值>2倍的microRNA分子。对筛选出的microRNA分子通过RT-qPCR方法,再次验证其在微血管内皮细胞中的表达水平。
3 统计学分析
所有实验数据均采用均数±标准误(mean±SEM)表示。利用SPSS 17.0软件对不同组数据分别进行平均值比较,两组间采用非配对t检验,多组间采用单因素方差分析(one-way ANOVA)进行数据统计,并用Tukey检验校正。以P<0.05为差异有统计学意义。
结 果
1 HSF1基因缺失加重压力超负荷下的左心室重构
心脏组织HE染色后进行CSA分析,结果显示TAC术后4周野生型C57BL/6小鼠的心肌细胞平均CSA要显著小于HSF1-/-小鼠(P<0.01),见图1A。心脏超声结果提示,TAC导致野生型小鼠的左室射血分数(left ventricular ejection fraction,LVEF)下降了17.7%(P<0.05);HSF1-/-小鼠的LVEF比野生型小鼠又进一步降低10.4%(P<0.05),见图1B;同时,左室舒张末内径(left ventricular end-dystolic diameter,LVEDD)的扩增程度在HSF1-/-小鼠组也更为明显(P<0.05),见图1C。心肌肥厚标志物的检测结果显示,β-MHC和ANP在假手术组间的mRNA表达没有差异,TAC致β-MHC和ANP的mRNA表达水平在HSF1-/-小鼠明显高于野生型小鼠(P<0.05),见图1D。同时,心脏组织的免疫组化染色显示,压力超负荷诱导下野生组CD31表达要明显高于HSF1敲除组,而在假手术情况下2种小鼠的心脏CD31表达均很低(图1E),提示压力超负荷可以诱导心脏CD31表达增加,通过血管新生改善心肌肥厚,而HSF1缺失抑制了这种保护作用。

Figure 1.HSF1 deficiency promoted pressure overload-induced cardiac hypertrophy. A: HE staining of the myocardial tissues (scale bar=20 μm) and determination of the cross-sectional area (CSA) of cardiomyocytes; B: determination of left ventricular ejection factor (LVEF); C: determination of left ventricular end-diastolic diameter (LVEDD); D: the mRNA levels of β-myosin heavy chain (β-MHC) and atrial natriuretic peptide (ANP) detected by RT-PCR; E: immunohistochemical staining for CD31 in myocardium (scale bar=20 μm). Mean±SEM.n=3.*P<0.05,**P<0.01vssham group;#P<0.05,##P<0.01vsWT group.
图1HSF1基因敲除促进压力超负荷下小鼠心肌肥厚
2 miR-195a-3p在HSF1敲除小鼠中表达显著增高
通过microRNA表达谱芯片我们筛到12个在HSF1-/-小鼠心脏中表达显著增高的microRNA,见图2A。因为HSF1参与血管新生信号通路激活,而血管新生障碍与心脏重构密切相关,因此我们进一步观察这些上调的microRNA与心肌微血管内皮细胞的关系,结果发现miR-34a-5p、miR-195a-3p、miR-208b-3p、miR-378a-5p和miR-451a在体外贴块培养的微血管内皮细胞中有不同程度的表达升高,而其他microRNA表达无差异,其中miR-195a-3p的表达显著增高(P<0.05),见图2B。
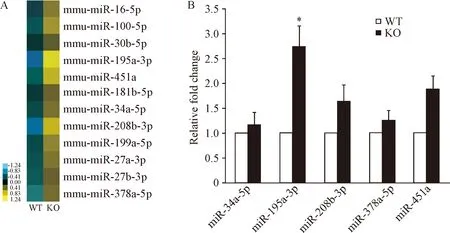
Figure 2.Screening for the microRNAs associated with angiogenesis-related signaling pathways which were upregulated in theHSF1 KO mice compared with the WT mice. A: the microRNAs up-regulated more than 2 folds inHSF1 KO mice were depicted by the heat map; B: the microRNAs highly expressed in microvascular endothelial cells were determined in the second-round screening using real-time PCR. Mean±SEM.n=3.*P<0.05vsWT mice.
图2在HSF1基因敲除小鼠模型中筛选与血管新生信号通路相关的microRNA
3 miR-195a-3p过表达抑制血管新生,促进微血管内皮细胞凋亡
血管新生功能实验结果显示,血管内皮生长因子(vascular endothelial growth factor, VEGF)诱导可促进内皮细胞快速生长和成管;而预先用miR-195a-3p mimic刺激细胞, VEGF介导细胞生长受抑,同时细胞成管数目明显减少(P<0.05),见图3A,TUNEL染色统计显示内皮细胞凋亡数目较阴性对照miRNA-NC干预组有显著增加(P<0.05),见图3B。通过Western blot 检测发现细胞内p53蛋白水平在miR-195a-3p mimic预处理后,较对照组表达升高2.24倍(P<0.05),较VEGF预处理组升高1.68倍(P<0.05);miRNA-NC干预后,p53的表达水平与对照组和VEGF诱导组比较均没有显著差异,见图3C。上述结果提示miR-195a-3p具有特异性抑制内皮细胞成管和促进内皮细胞凋亡的作用。
4 miR-195a-3p通过抑制 AMPKα2的表达,减少血管新生
通过生物信息软件TargetScan 6.2分析可能受miR-195a-3p调控的靶基因,筛选出预测综合得分排序较高、且涉及血管新生信号通路的基因——PRKAA2,其编码蛋白为AMPKα2。双萤光素酶报告基因测定结果表明,位于AMPKα2基因序列的5736~5757区域发生无义突变后,miR-195 mimic抑制AMPKα2转录的效应明显增强,见图4A。我们进一步用Western blot检测miR-195 mimic对于心肌微血管内皮细胞的AMPKα2蛋白表达影响,结果发现转染miR-195 mimic明显抑制AMPKα2的表达,而miR-NC没有抑制作用,见图4B。同时,血管新生因子CD31和VEGF的蛋白表达水平也被miR-195 mimic显著抑制,而miR-NC对于CD31和VEGF的抑制作用不明显,见图4C。这些结果提示miR-195a-3p可能通过抑制AMPKα2的表达,干预微血管内皮细胞的血管新生。
讨 论
在本研究中,我们发现HSF1基因缺失导致血管再生障碍,加速了TAC诱导下的心肌肥厚向心衰发展。HSF1缺失的小鼠心脏microRNAs表达谱与野生型小鼠有明显差别,其中miR-195a-3p在心脏微血管内皮细胞中表达明显升高; miR-195a-3p通过3’-UTR靶向抑制AMPKα2,同时miR-195a-3p过表达也抑制了心脏微血管内皮细胞的CD31和VEGF的表达。以上这些发现提示miR-195a-3p靶向抑制AMPKα2可能是其抑制血管新生的机制;通过表观遗传学的修饰和调控,可能是预防和治疗高血压等压力超负荷等应激引起心脏重构的重要措施。
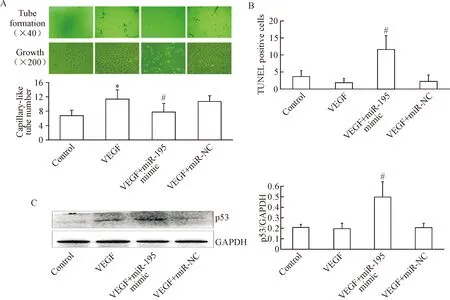
Figure 3.miR-195a-3p suppressed the growth and tube formation of microvascular endothelial cells. The cells were stimulated by vascular endothelial growth factor (VEGF) alone, VEGF+miR-195 mimic or VEGF+negative control microRNA (miR-NC). A: the growth and tube formation of the cells were observed, and the numbers for capillary-like tubes were determined; B: the apoptosis of the cells was detected by TUNEL assay; C: the protein expression of p53 was detected by Western blot. Mean±SEM.n=3.*P<0.05vscontrol group;#P<0.05vsVEGF group.
图3miR-195a-3p促进微血管内皮细胞凋亡和抑制血管再生
我们课题组既往报道了HSF1基因缺失会加重压力超负荷下引起的小鼠心肌肥厚和心力衰竭的体征出现[3]。在该研究中,我们主要发现HSF1过表达可以抑制压力超负荷下心脏p53的增加,并促进血管新生因子VEGF和Ang1的表达,而HSF1敲除小鼠会出现严重的血管新生障碍。因此,我们推测HSF1基因敲除引起的内皮细胞凋亡和血管新生不足可能是促使压力超负荷下HSF1-/-小鼠严重缺血缺氧、心肌细胞肥大、加速心衰的主要原因。但是,HSF1缺失介导心脏血管新生障碍的病理分子机制仍不清楚。
在本研究中,我们从表观遗传学的角度分析,HSF1缺失引起的microRNA表达谱差异造成血管新生障碍的可能性。芯片数据提示,HSF1缺失的C57小鼠心脏microRNAs的表达谱不同于正常对照小鼠,并且有12个microRNA分子显著上调;通过在心肌微血管内皮细胞中进一步验证,发现miR-195a-3p是HSF1缺失后在细胞中上调最明显的分子。miR-195上调可以抑制肿瘤细胞的增殖,其机制是:DNA倍增是促使肿瘤细胞由G1期快速向S期过渡的关键,而miR-195可以靶向抑制这一细胞时期的多个编码基因如CCND1、CDK6和EGF2等,从而促进肿瘤细胞的凋亡[8]。因此,我们认为在心肌微血管内皮细胞中的miR-195a-3p表达增高,可能也是通过调控内皮细胞中的一些周期基因,造成内皮细胞的凋亡并影响到内皮血管再生功能。通过TargetScan软件分析,我们确实找到这些细胞周期基因可能是miR-195靶点的证据,但是我们找到一个更为关键的靶基因PRKAA2——AMPKα2的编码基因。双萤光素酶报告基因测定结果证实miR-195a-3p可以作用于PRKAA2基因的3’-UTR区域。由于AMPKα2在心脏血管新生代谢中发挥非常重要的作用[9-14],例如,AMPKα2缺失可以抑制下游UCP2、VEGF等蛋白的表达,导致内皮功能紊乱和血管新生障碍[9-10];而AMPKα2激活则可以磷酸化eNOS并促进VEGF依赖的血管再生和血管舒张[11-12]。此外,AMPKα2激活还可以逆转老化微血管内皮细胞的血管新生功能。在AMPKα2敲除小鼠中,压力超负荷可以加重左室肥厚和心功能紊乱[13]。因此,我们推测miR-195可能不仅仅参与抑制细胞周期基因,更重要是直接抑制心脏血管新生通路上的关键基因,从而加速了压力超负荷下血管新生障碍和心脏重构。
Figure 4.miR-195a-3p inhibited AMPKα2 and angiogenesis-related genes. A: sequence alignment of mouse miR-195a-3p with 3’-UTR of AMPKα2 and the result of dual-luciferase reporter assay; B: the protein level of AMPKα2 was detected by Western blot; C: the expression and quantitative analysis of CD31 and VEGF were performed by Western blot. Mean±SEM.n=3.*P<0.05vscontrol group.
图4miR-195a-3p通过作用于AMPKα2抑制血管新生相关信号通路
在本研究中,我们证实了miR-195a-3p过表达不仅有效抑制了心脏微血管内皮细胞中AMPKα2的表达,同时也抑制了血管新生相关基因CD31和VEGF的表达,提示miR-195a-3p对AMPKα2的靶向抑制可能与心脏血管再生功能紊乱有一定关系。值得注意的是,在本研究中miR-195a-3p的升高与HSF1的缺失有密切关系。荷兰学者van Rooij等[15]在一项研究中发现,miR-195心肌特异性高表达小鼠会出现快速心衰表现,在繁育该品系的小鼠出生第2周后即出现死亡;进一步的深入研究确证,在心脏发育过程中如果miR-195表达异常增高,可能导致先天性心脏病,包括室上间隔缺损和严重的心室发育不全等症状。而miR-195异常增高引起的这些心脏病理特征,在HSF1敲除小鼠也发现类似现象[3, 16]。结合本实验结果,提示 HSF1和miR-195存在“cross-talk”或者信号通路的上下游关系,而在压力超负荷下,HSF1的表达可以有效抑制机械应力下心脏miR-195a-3p的增加,从而逆转miR-195a-3p对AMPKα2的抑制,并可能因此激活下游CD31和VEGF等血管新生相关因子,促进内皮细胞的血管新生能力,改善压力超负荷下的心肌重构。
综上所述,本研究通过构建小鼠压力超负荷模型,揭示了HSF1基因的缺失可导致心脏miR-195a-3p的表达异常升高,从而抑制下游AMPKα2的表达,阻滞代偿性的血管新生,最终加速了压力超负荷下心脏重构和心力衰竭的发生。
