酱香型白酒大曲和糟醅的细菌多样性分析
2019-03-11戴奕杰李宗军田志强
戴奕杰,李宗军*,田志强
(1.湖南农业大学食品科技学院,湖南 长沙 410128;2.贵州省产品质量监督检验院,贵州 贵阳 550016)
白酒是中国的传统酒类,是世界六大蒸馏酒之一。其中,酱香型白酒以其“酱香突出、优雅细腻、柔绵醇厚、回味悠长”的独特风味,受到广大消费者的青睐,驰名中外[1-2]。研究者认为,特殊的风格离不开独特的酿造工艺和酿造方法:“四高二长、一大一小”,即高温制曲、高温堆积、高温发酵、高温馏酒、生产周期长、贮存时间长、大用曲量、多轮次(发酵)取酒,每一个环节在酱香酒的酿造生产中都是不可或缺的,时间以1 a为1 个生产周期[3-6]。
白酒中最重要且研究最多的微生物类群是细菌,其主要来自于高温制曲和堆积工序。方心芳先生认为,酱香白酒的典型-茅台曲是一种细菌曲[7]。固态发酵的微生物变化及生化反应主要发生在高温制制曲、堆积发酵工序。高温制曲中,细菌占绝对优势,有90%以上的数量,以获得所需香味物质[3,8-9]。高温堆积又网罗了空气环境中的有益微生物,相当于“二次制曲”[10-12]。细菌及其代谢产物在高温操作过程中强化了糟醅自身形成酱香的原动力,促进酱香物质的进一步生成,研究最多的枯草芽孢杆菌和地衣芽孢杆菌被认为是利用糖类和氨基酸促成美拉德反应而大量产生呈香物质[13]。窖池发酵是将前者的代谢产物进行积累利用的过程,产酸细菌(乳酸菌、醋酸菌等)成为主力,产乙醇和二氧化碳的同时,也是酸类、酯类物质(乳酸、乙酸、乳酸乙酯、乙酸乙酯等)形成的基础[14]。
高温大曲是优质酱香白酒重要的糖化剂、发酵剂和生香剂,具有原料易得、工艺简单、功能性强的特点[2]。大曲在白酒酿造中有着举足轻重的作用,其中栖息着大量微生物种属,如细菌、霉菌及酵母等[15]。微生物成为决定大曲品质的核心因素。20世纪80年代后期,人们对传统固态发酵白酒的再认识,使得大曲微生物的研究逐渐进入高潮[16]。王忠彦[17]、施安辉[18]、廖建民[19]等利用传统可培养方法对大曲中的微生物进行探索和研究,揭示了大曲微生物优势菌群的种属分布,得到的微生物分为霉菌、细菌、酵母菌等,数量上以细菌菌群最多,霉菌次之,酵母菌则较少。聚合酶链式反应-变性梯度凝胶电泳(polymerase chain reaction-denaturing gradient gel electrophoresis,PCR-DGGE)技术于21世纪初运用到大曲微生物的研究中,使人们可以更进一步认识曲酒中的微生物。Chen Tingtao等[20]酱香酒大曲中的细菌主要有芽孢杆菌、乳酸细菌、醋酸细菌等,它们是酒曲蛋白酶和淀粉酶的重要来源,对后续发酵和风味成型均具有重要影响[21]。
糟醅堆积发酵阶段又称为“二次制曲”[1,22],在高温环境下,高温大曲的曲药微生物能在糟醅上增殖、发酵,同时高温堆积网罗、富集环境空气中的微生物,生成酱香或酱香前体物质,为入窖发酵创造条件[23-24]。在堆积过程中,细菌和霉菌数量减少,而酵母快速生长,这一过程极大增加了酵母数量,使得窖池发酵正常进行。窖池发酵阶段是产酒的过程,此时随着发酵时间的延长,微生物数量下降,酵母和霉菌在发酵后期无法检测到[25-26]。由于堆积温度在50 ℃左右,极端酿造环境中的微生物多属于中温嗜热菌[27]。其中,细菌以芽孢杆菌属为主,包括地衣芽孢杆菌、巨大芽孢杆菌、枯草芽孢杆菌、同时还有葡萄球菌属、土壤杆菌属等。正是由于酱香型白酒酿造过程中微生物的多样性特点,共同推动了产品生产的顺利进行,对酱香白酒品质、风味的形成具有重要作用。
Illumina HiSeq 2500测序系统采用pair-end边合成边测序技术,对环境微生物的分析已经趋向成熟,相关数据库中的菌种分类也更加全面具体。本研究采用高通量测序技术(HiSeq测序)及生物信息学方法,对大曲和糟醅样本中的细菌多样性分析,以期更全面地解析酱香型白酒酿造过程中微生物群落结构特点和动态变化规律,以及不同工艺阶段菌群组成的差异[28]。
1 材料与方法
1.1 材料与试剂
1.1.1 大曲样本采集
本实验样本是跟踪酱香型白酒生产企业实地生产过程中,选取酱香高温制曲3 个阶段的曲块,采用5 点取样法:以曲块对角线的中点作为中心抽样点,再于对角线上选择4 个与中心样点距离相等的点作为抽样点,5 个样点取相同体积的曲块。将上述采样位点的样品直接破碎并混匀后,分装于无菌袋封装标记,于-80 ℃冰箱中保存。具体采样信息见表1。

表1 高温大曲样本信息Table 1 Information about samples of high-temperature Daqu
1.1.2 糟醅样本采集

表2 发酵酿酒糟醅样本信息Table 2 Information about samples of fermented grains
按照传统酱酒的酿造工艺,采用上述准备的大曲,在酒厂进行酱酒的酿造。选取整个酱香白酒发酵工艺的下沙、造沙和7 轮次发酵过程的高温堆积、窖池发酵工序的糟醅,具体取样时间如表2所示,采用如图1、2中的位置对高温堆积和窖池发酵糟醅进行取样,每个取样的量保持一致。将每次取样点糟醅直接混匀后分装于无菌袋封装标记,于-80 ℃冰箱中保存。
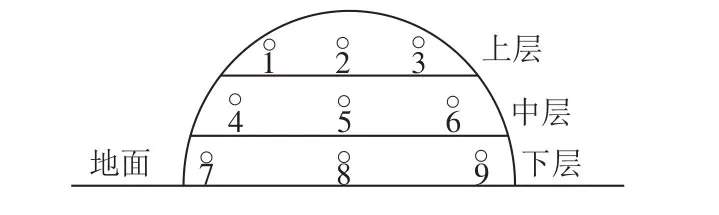
图1 高温堆积过程取样点位示意图Fig. 1 Schematic diagram of sampling points during high-temperature stacking fermentation

图2 窖池发酵糟醅取样示意图Fig. 2 Sampling points of fermented grains in the pit
1.1.3 试剂
DNA提取试剂盒 美国MP Biomedicals公司; HiSeq Rapid SBS Kit v2测序试剂盒(FC-402-4023 500 Cycle)及文库构建TruSeq DNA PCR-Free Sample Prep Kit(FC-121-3001/3003) 美国Illumina公司。
1.2 仪器与设备
Qubit 2.0荧光定量仪 美国Thermo Fisher公司;NanoVue系统 美国GE Healthcare公司。
1.3 方法
1.3.1 样品预处理
称取7 g大曲和糟醅样品,用20 mL灭菌后的0.1 mol/L磷酸盐缓冲液(phosphate buffer saline,PBS)悬浮,加入玻璃珠,旋涡振荡5 min。300×g离心5 min,取上清液,沉淀用PBS重复洗涤3 次,离心后收集上清液。全部上清液于10 000×g离心15 min,弃去上清液,收集细胞沉淀。再用5 mL PBS洗3 次,每次以10 000×g离心15 min,收集沉淀。最后将沉淀重新悬浮于2 mL PBS中,-20 ℃保存[29]。
1.3.2 样品DNA提取及16S rDNA测序分析
采用环境样本D N A提取试剂盒进行基因组D N A抽提后,使用0.8%的琼脂糖凝胶电泳检测DNA。使用细菌的16S rDNA通用引物对样品所提取的DNA进行扩增,16S rDNA V4可变区引物515F(5’-GTGCCAGCMGCCGCGGTAA-3’)和806R(5’-GGACTACHVGGGTWTCTAAT-3’)[30]。每个样本进行3 个重复,每个PCR终止于线性扩增期,PCR结束后将同一样本的PCR产物混合后用2%琼脂糖凝胶电泳检测,检测条件5 V/cm、20 min,使用OMEGA胶回收试剂盒切胶回收PCR产物,Tris-EDTA缓冲液洗脱回收目标DNA片段。将PCR回收产物用Qubit 2.0或NanoVue系统进行检测定量,之后按照每个样本的测序量要求,进行相应比例的混合。使用TruSeq DNA PCR-Free Sample Prep Kit进行文库构建。使用HiSeq Rapid SBS Kit v2测序试剂盒进行测序。
1.4 高通量测序数据统计分析
对于双端测序得到的PE reads首先使用FLASH[31]进行拼接,根据Barcode区分不同来源的样本。对序列质量进行质控,在去除低质量碱基及接头污染序列等操作过程后完成数据过滤,得到可供后续分析的高质量有效序列。
使用QIIME[32]等软件在97%的相似性水平上利用UPARSE算法[33]进行操作分类单元(operational taxonomic units,OTU)的聚类,并挑选出OTU的代表性序列。
使用Uchime[34]去除嵌合体,并采用Silva等数据库[35]进行物种分类信息的划分,同时去除注释为叶绿体、线粒体以及非细菌或古菌界的OTU。
使用PyNAST算法[36]对代表性序列进行比对并过滤,然后使用FastTree[37]重构建进化树。从OTU表中去除在进化树重构建过程中被过滤掉的OTU,重抽样OTU表使每个样品具有相同的序列数。
基于上述结果在群落组成水平和系统发育水平上使用QIIME和R等软件对样本进行Alpha多样性分析、Beta多样性分析和群落结构的差异性分析等,在物种水平上进行物种的差异分析和相关分析等。
2 结果与分析
2.1 酱香型白酒大曲的细菌多样性分析
2.1.1 细菌群落多样性指数
使用UPARSE算法在97%的相似性水平上进行OTU的聚类,筛选出OTU的代表性序列,并比对数据库进行物种分类信息的划分。使用QIIME对样本进行Alpha多样性分析,包括丰富程度等级(Rank-Abundance)曲线,以及以Observed、Shannon及Chao1指数绘制的稀疏曲线。
Rank-Abundance曲线是分析多样性的一种方式。构建方法是统计单一样本中,每个OTU所含的序列数,将OTU按丰度(所含有的序列条数)由大到小等级排序,再以OTU等级为横坐标,以累积序列丰度为纵坐标作图。Rank-Abundance曲线可用来解释多样性的两个方面,即物种多度和物种均匀度。在水平方向,物种的多度由曲线的宽度来反映,物种的多度越高,曲线在横轴上的范围越大;曲线的形状(平滑程度)反映了样本中物种的均度,曲线越平缓,物种分布越均匀。从图3可看出,物种分布较为均匀。

图3 大曲16S rDNA Rank-Abundance曲线Fig. 3 Rank-Abundance curves for bacterial 16S rDNA from Daqu

图4 大曲16S rDNA多样性指数分析Fig. 4 16S rDNA diversity analysis of bacteria from Daqu
稀疏曲线是从样本中随机抽取一定数量的个体样本,统计这些个体所代表的物种数目,以图示反映样本中物种的丰富程度。当曲线趋于平缓时可认为测序深度已经基本覆盖到样品中所有的物种。样本曲线的延伸终点的横坐标位置为该样本的测序数量,如果曲线趋于平坦表明测序已趋于饱和,增加测序数据无法再找到更多的OTU;反之表明不饱和,增加数据量可以发现更多OTU。图4结果显示,高温制曲阶段,细菌的微生物丰富度较好,本实验的测序量也能够较好地反映出样本内的微生物信息。表3丰度指数显示,随着发酵时间的推移,大曲的多样性指数(Shannon:3.40)、分类OTU(Observed:360)、物种丰度指数(Chao1:581)均趋于稳定。

表3 基于16S rDNA测序高温大曲Alpha多样性指数Table 3 Alpha diversity indexes based on 16S rDNA sequencing of bacteria from high temperature Daqu
2.1.2 细菌群落结构分析

图5 门分类水平上的细菌菌群Fig. 5 Bacterial community structure at phylum level

图6 属分类水平上的细菌菌群Fig. 6 Taxonomic analysis of bacteria at genus level
将每个OTU与数据库进行比对,选取置信度阈值,即相似度在97%以上的序列进行物种分类,得到每个OTU的分类水平,即门、纲、目、科、属分类水平。分析高温制曲3 个时期样本的细菌菌类,从图5、6可以看出,高温制曲阶段,占据主导地位的细菌菌群为厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、拟杆菌门(Bacteroidetes)4 类菌门;随着曲药贮存时间的延长,Firmicutes和Bacteroidetes的菌群数量逐渐下降,而Proteobacteria、Actinobacteria的数量则呈现上升趋势。根据测序结果,在大曲中共鉴定出18 个门167 个属的细菌微生物。其中,优势菌属有Clostridium sensu stricto1、Escherichia-Shigella、Lactobacillus、Streptococcus、Actinobacillus、Bifidobacterium、Peptoclostridium、Mycoplasma、Citrobacter等,诸如Escherichia-Shigella、Actinobacillus、Bifidobacterium、Citrobacter等细菌随贮存时间的延长而增加,Clostridium sensu stricto1、Mycoplasma等表现为先上升后下降,而Lactobacillus、Peptoclostridium等无明显变化,Streptococcus等细菌呈现下降趋势。高温制曲阶段,由于高温条件下,保留大量的嗜热性细菌,研究者认为酱香物质是来源于曲药,即高温制曲产物。本实验中梭菌属、芽孢杆菌、乳酸菌等占主导优势,它们能够分泌蛋白酶、淀粉酶等水解原料,产生发酵性糖和氨基酸,为发酵酿酒过程中美拉德反应的进行提供大量原料。
2.2 酱香型白酒发酵过程中糟醅的细菌多样性分析
2.2.1 细菌群落多样性指数

图7 糟醅16S rDNA多样性指数分析Fig. 7 16S rDNA diversity analysis of bacteria from fermented grains
传统的白酒酿造是开放式-半开放式的生产,环境空气、车间、工具、发酵设备、参与人员等都附着着丰富的微生物群,通过不断的生长繁殖进入到糟醅之中,因此,堆积发酵也有“二次制曲”之称。分析“四高”中的高温堆积和窖池发酵阶段细菌微生物群落结构,从下沙环节开始到第7轮次酒起窖为止,共采集16 个糟醅样本进行测序分析,图7结果显示,堆积发酵和窖池发酵阶段,曲线较为平缓,说明细菌的微生物分布均匀且物种组成丰富。表4与图8显示,OTU个数均达270以上;随着发酵时间的推移,发酵过程的多样性指数(Shannon)除了第3轮次下窖样本较低,其他样本均在3.0以上;物种丰度指数(Chao1)基本呈现堆积发酵(下窖)样本低,窖池发酵(起窖)样本高的变化。

表4 基于16S rDNA测序糟醅Alpha多样性指数Table 4 Alpha diversity indexes based on 16S rDNA sequencing of bacteria from fermented grains

图8 糟醅16S rDNA多样性指数分析Fig. 8 16S rDNA diversity analysis of bacteria from fermented grains
2.2.2 细菌群落结构分析
从图9、10可看出,发酵时期,占据主导地位的细菌菌群为Firmicutes、Proteobacteria、Actinobacteria、Bacteroidetes 4 类;随着发酵时间的推移,Firmicutes前期下降,中期从第2轮次起窖开始菌类上升,后期的第6轮次起窖种类下降,其中,第3轮次下窖占比达到最高值(0.817 851 959)、第4轮次起窖最低(0.400 217 707);Proteobacteria变化趋势与前者相反;Actinobacteria、Bacteroidetes则无明显变化规律。对OTU分类结果进行分析统计,发酵阶段共鉴定出22 个门和221 个属,相对丰度较高菌属为:Escherichia-Shigella、Lactobacillus、Clostridium sensu stricto1、Streptococcus、Actinobacillus、Bifidobacterium、Peptoclostridium、Citrobacter等。糟醅细菌中Escherichia-Shigella、Lactobacillus在发酵前期菌数先升高,中期从第2轮次开始下降,第3轮次下窖时达到最低,后期逐渐上升。Clostridium sensu stricto1、Streptococcus菌群丰度变化规律复杂,呈现先下降后上升的趋势,值得一提的是Clostridium sensu stricto1菌种在第3轮次下窖时达到最高。Actinobacillus、Bifidobacterium、Peptoclostridium、Citrobacter虽占比不及前三者,但变化较为稳定。
发酵酿酒过程分为高温堆积和窖池发酵两部分:高温堆积阶段将粮醅或蒸馏后的糟醅在晾堂摊凉、拌曲后堆成的圆堆,进行堆积发酵至顶温;窖池发酵是加入曲的粮糟在窖池内糖化,依赖于窖池中各种微生物群落的功能和作用完成酿造过程。本实验发酵酿酒工艺中,细菌微生物相对丰度较高,优势细菌Escherichia-Shigella、Lactobacillus随轮次的增加,表现为前期堆积发酵(下窖)高、窖池发酵(起窖)低,后期窖池发酵(起窖)高的变化趋势;而Clostridium sensu stricto1、Streptococcus等表现相反变化;说明部分芽孢杆菌属及乳杆菌属在窖泥种代谢旺盛,为下窖提供前体物质,并影响着轮次酒的风格形成。

图9 门分类水平上的细菌菌群Fig. 9 Bacterial community structure at phylum level

图10 属分类水平上的细菌菌群Fig. 10 Bacterial community structure at genus level
2.3 大曲和糟醅的细菌多样性比较
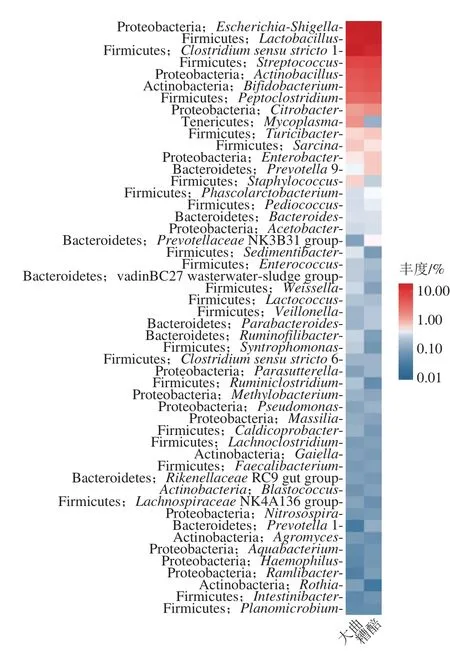
图11 属分类水平细菌的丰度类群热图Fig. 11 Heat map of relative abundance of bacteria at genus level
特定的生产工艺导致了特定的微生物组成和动态变化,由于高温制曲和堆积发酵时期存在的微生物群落存在差异,其中物质代谢、能量代谢的结果也有所不同,导致了酱香型白酒特征风味的差异。本节针对大曲和糟醅中的细菌群落结构进行比较分析,包括聚类分析、韦恩分析、网络Network分析,以期了解其中优势菌类的差异性和不同时期细菌微生物间的相互作用。
从图11可知,无论是高温制曲时期还是发酵时期,占据主导地位的细菌菌群为Firmicutes,其中占比较多的属是Clostridium sensu stricto1、Lactobacillus、Streptococcus;其次是变形菌门Proteobacteria,其中占比较多的属是Escherichia-Shigella、Actinobacillus;然后是放线菌门Actinobacteria,其中Bifidobacterium占比最多。另外,丰度较低的细菌菌类中Mycoplasma、Sarcina、Staphylococcus在大曲中的丰度高于糟醅,Citrobacter、Turicibacter、Enterobacter、Prevotella9在大曲中的丰度低于糟醅样本。

图12 样本OTU韦恩图Fig. 12 Wayne diagram for bacterial OTUs from Daqu and fermented grains
图12 显示,大曲和糟醅的OTU中有98.66%的序列占比形成交集,而糟醅有342 个OTU独有,相比之下,大曲只有28 个OTU独有。
3 结 论
通过采集大曲和糟醅不同发酵时间的样本,提取细菌DNA并根据16S rDNA V4多变区序列的特异性引物进行PCR扩增,随后利用Illumina HiSeq 2500测序技术对扩增产物进行高通量测序。得到的测序数据使用生物信息学方法进行微生物多样性分析,包括OTU、Alpha多样性分析、群落组成分析等。结果显示:酱香型白酒的高温制曲、堆积发酵和窖池发酵的微生物菌群在数量及结构分布上具有多样性,菌群种类基本一致。两个阶段的优势菌门均为Firmicutes、Proteobacteria、Actinobacteria、Bacteroidetes 4 类。随着发酵时间的推移,各轮次酒糟醅中微生物群落呈现一定的变化规律,大曲样本的细菌菌类变化与贮存时间有关,而糟醅微生物随窖内发酵及高温堆积的工艺而不断变化。随着温度和水分达到相对平衡,细菌群落也逐渐得到了调整,形成单一菌类为主导的环境,更有利于酱香型白酒的酿造。
制曲和制酒阶段的生态环境之间存在差异,导致微生物优势菌不同:大曲相对丰度较高的菌属为Clostridium sensu stricto 1、Escherichia-Shigella、Lactobacillus、Streptococcus、Actinobacillus、Bifidobacterium、Peptoclostridium、Mycoplasma、Citrobacter等。糟醅的优势菌属为Escherichia-Shigella、Lactobacillus、Clostridium sensu stricto 1、Streptococcus、Actinobacillus、Bifidobacterium、Peptoclostridium、Citrobacter。韦恩分析显示,2 个阶段的微生物菌落既有交集也有各自特有的菌类,结合热图比较发现,存在一些细菌菌类丰度上的差异,如Mycoplasma、Sarcina、Staphylococcus在大曲中的丰度高于糟醅,柠檬酸杆菌属Citrobacter、Turicibacter、Enterobacter、Prevotella 9低于糟醅样本。
