3个黑龙江烟区烟草花叶病毒分离物的全基因组序列测定与分析
2019-03-11姜瀚林郭兆奎刘永中万秀清刘文涛李现道李向东
姜瀚林,郭兆奎,刘永中,万秀清,刘文涛 ,李现道,李向东
1 山东农业大学,植物保护学院植物病毒学研究室,山东泰安岱宗大街61号 271018;
2 黑龙江省烟草科学研究所,黑龙江哈尔滨哈药路17号 150076;
3 山东临沂烟草有限公司,山东省临沂市兰山区北城新区智圣路3号 276003;
4 山东省烟草研究院,山东济南舜华东路13号 250098
烟草花叶病毒(Tobacco mosaic virus,TMV)是烟草花叶病毒属(Tobamovirus)的代表种,是危害烟草、番茄和马铃薯的主要病毒。TMV侵染烟草可引起花叶及疱斑等症状,严重影响烟叶产量及品质。TMV基因组为正义单链RNA,5′端有m7G5′pppG帽子结构,3′端有tRNA-like结构,基因组全长6395核苷酸(nt)左右[1-3]。
明确TMV分子株系对指导病毒早期检测及防治有重大意义。谢扬军等[4]对国内8个主要产烟省的TMV分离物进行株系划分,证明我国TMV株系划分存在地理隔离。杨恭等[5]测定了TMV中国U1株系分离物及弱毒番茄分离物的全基因组序列。邵碧英等[6]测定了福建TMV普通株系及两个弱毒突变体的全基因组序列。庞小静[7]等测定了TMV山西分离物的全基因组序列。目前还没有关于黑龙江TMV烟草分离物全基因组序列测定及分析的报道。本文测定了3个黑龙江烟草TMV分离物的全基因组序列,并与GenBank中26个TMV分离物进行了一致率、重组、选择压力和系统发育等分析,为黑龙江烟区TMV检测及抗病育种提供理论依据。
1 材料与方法
1.1 材料
发病烟草样品采自黑龙江省哈尔滨市和牡丹江市,3个样品分别命名为HEB1、HEB2和MDJ。大肠杆菌DH5α由本实验室保存。植物总RNA提取试剂盒Transzol、DNA凝胶回收试剂盒等均购自北京全式金公司;TaqDNA聚合酶购自南京诺唯赞公司,M-MLV反转录酶、pMD18-T克隆载体、LATaqDNA聚合酶和末端转移酶购自TaKaRa公司; Super ScriptTMⅣ反转录酶购自Invitrogen公司。其他生化试剂及普通化学试剂均为进口或国产分析纯。
1.2 实验方法
1.2.1 扩增策略及引物合成
使 用DNAMAN(version7)对GenBank中 的TMV序列进行比对,选择保守片段设计引物,分两段进行全序列扩增。引物序列详见表1。

表1 TMV全基因组扩增引物Tab.1 Oligonucleotide primers used to amplify the complete genome of TMV
1.2.2 植物总RNA提取、RT-PCR扩增及5′RACE
利用Transzol试剂盒提取植物总RNA,M-MLV反转录酶进行反转录,分两段扩增5′端以外的TMV基因组片段。依照Elizabeth等[8]的5′ RACE扩增TMV 5′端基因组。
1.2.3 克隆及序列测定
PCR产物经1%琼脂糖凝胶电泳分离后,紫外灯下切取3498 bp和3470 bp处胶条。使用EasyPure Quick Gel Extraction Kit回收PCR产物并连接pMD18-T载体,转化大肠杆菌(Escherichia coli)DH5α感受态细胞后,蓝白斑筛选挑取白色单菌落,PCR验证阳性后选取3个克隆送北京华大基因公司测序。
1.2.4 核苷酸序列一致率分析
使用DNASTAR软件包的Seqman对序列进行拼接,通过MegAlign与GenBank中26个TMV分离物(表2)序列进行比对,分析3个分离物全基因组在氨基酸和核苷酸水平与参比序列的一致率。

表2 本文分析用的29个TMV分离物序列号及来源Tab.2 Accession number, host and geographical origin of the 29 TMV isolates analyzed in the paper
续表2
1.2.6 系统发育分析
以ToMV queensland分离物为外组,选取GenBank中26个TMV分离物,通过MEGA7中 CLUSTAL W模块进行序列对比,以邻近法(Neighbor-Joining,NJ)构建系统进化树,自展值设置为1000并去掉小于50%的分支。同时使用MEGA7计算组内距离和组间距离。
1.2.5 重组分析
将序列用 CLUSTAL X1.81软件进行比对,使用RDP(version4.95)软件包进行重组分析。通过 RDP、GENECONV、BOOTSCAN、MAXCHI、CHIMAERA、SISCAN 和 3Seq 七种算法分析。当四种以上算法支持重组,且P<1.0×10-6时,该分离物可认定为存在重组。
1.2.7 选择压力分析
使用MEGA7中Pamilo-Bianchi-Li算法,分别计算各蛋白的dN/dS值,若dN/dS>1,说明存在正选择;若dN/dS=1,说明存在中性选择;若dN/dS<1,说明存在负选择。
2 结果与分析
2.1 分离物HEB1、HEB2和MDJ基因组结构
HEB1、HEB2和MDJ 3个分离物基因组RNA长度均为6395 nt(GenBank登录号分别为MH595919、MH595920和MH595921)。分离物HEB1基因组RNA A、C、G、U的含量为别为29.1%、19.0%、24.1%和27.7%,HEB2为29.2%、19.0% 、24.1%和27.8%,MDJ为29.1%、18.9%、24.1%和27.8%。
3个分离物基因组均包含4个开放阅读框(ORF)。ORF1起于69 nt,止于3419 nt,编码分子量为126 kDa的蛋白,终止密码子TAG可被通读至4919 nt形成ORF2,编码分子量183 kDa的通读蛋白,两种蛋白共同行使依赖于RNA的RNA聚合酶功能(RNAdependent RNA polymerase,RdRp)。ORF3起 于4903 nt,止于5709 nt,编码分子量30 kDa的移动蛋白(movement protein,MP)。ORF4起于5712 nt,止于6191 nt,编码分子量17.6 kDa的外壳蛋白(coat protein,CP)(图1)。
2.2 核苷酸和氨基酸一致率分析
在全基因组水平上(表3),HEB1分离物基因组与Beipiao分离物的核苷酸一致率最高,为98.9%,与petTW分离物一致率最低,为85.8%。其中,ORF1/2与Beipiao分离物一致率最高,为99.4%,与Ohio V分离物一致率最低,为86.2%;ORF3与HEB2分离物一致率最高,为98.0%,与petTW分离物一致率最低,为81.6%;ORF4与MDJ分离物一致率最高,为99.3%,与petTW分离物和Ohio V分离物最低,同为89.1% 。HEB2分离物基因组与Chuxiong1分离物的核苷酸一致率最高,为97.1%,与petTW分离物一致率最低,为85.4%。其中,ORF1/2、ORF3和ORF4均与Chuxiong1分离物一致率最高,分别为97.0%、96.8%和97.9%,ORF1/2与Ohio V分离物一致率最低,为86.0%,ORF3和ORF4则与petTW分离物一致率最低,分别为81.1%和88.4%。MDJ分离物基因组与Beipiao分离物的核苷酸一致率最高,为99.6%,与petTW分离物一致率最低,为85.8%。其中,ORF1/2、ORF3和ORF4均与Beipiao分离物一致率最高,分别为99.6%、99.4%和99.8%;ORF1/2与Ohio V分离物一致率最低,为86.2%,ORF3与petTW分离物一致率最低,为81.3%,ORF4与petTW分离物和Ohio V分离物最低,同为88.9%。
在氨基酸水平上(表4),HEB1分离物183 kDa和MP均与MDJ分离物的氨基酸一致率最高,分别为98.8%和99.6%,183 kDa与petTW分离物和Ohio V分离物氨基酸一致率最低,均为95.5%;MP与petTW分离物氨基酸一致率最低,为86.8%。HEB2分离物183 kDa和MP均与Chuxiong1分离物氨基酸一致率最高,分别为99.0%和97.5%;183 kDa与petTW分离物和Ohio V分离物一致率最低,均为95.7%,MP则与petTW分离物一致率最低,为85.5%。MDJ分离物183 kDa与Beipiao分离物和TMV152分离物一致率最高,为99.8%,与Ohio V一致率最低,为96.2%;MP与Xingren1、Shenyang、Yihan1、Hechi和Songtao1等5个 分 离物一致率最高,均为99.6%,与分离物petTW一致率最低,为87.2%。三个分离物CP的氨基酸序列与其他分离物CP的氨基酸一致率相同,除petTW、Yihan1、Zigui、Rakkyo和Ohio V五个分离物之外,其他分离物一致率均为100%;与分离物Ohio V一致率最低,为97.5%。
2.3 分离物HEB1、HEB2和MDJ基因组重组分析
使 用CLUSTAL W将HEB1、HEB2和MDJ与其他26个TMV分离物序列比对,利用RDP分析可能重组情况。结果表明,所用的7种软件都检测到HEB1存在重组事件(表5),且其P值远远小于<1.0×10-6,说明其基因组存在明显重组。HEB1基因组4780 nt -5462 nt来自HEB2(minor parent),其它部分来自MDJ(major parent)(图2)。

图2 HEB1重组模式示意图Fig.2 Recombination pattern of HEB1
2.4 系统进化关系
系统进化分析结果表明,29个TMV分离物聚为3个组(图3)。I(U1)组最大,包括24个分离物,以U1分离物为代表。该组又可以分为2个亚组,其中包括本研究中的HEB1、MDJ两个分离物以及福建U1株系分离物(FujianU1)、TMV152等共13个中国分离物聚为一个亚组,说明中国大部分TMV基因组具有一定保守性。来自中国蚕豆、西德和美国的11个分离物形成第2亚组。第II组包括HEB2、云南chuxiong1和日本Rakkyo三个分离物,以Rakkyo分离物为代表。台湾petTW与美国Ohio V分离物聚为III(Ohio)组。三个组中都有中国分离物,说明中国TMV分离物具有更高的遗传多样性。
为验证分组的可信度,计算组内距离和组间距离(表6)。结果表明I(U1)组内遗传距离为0.009,II(Rakkyo)和III(Ohio)组内遗传距离较大,分别为0.021和0.022,但组间距离大于组内距离,证明该系统进化树可信。
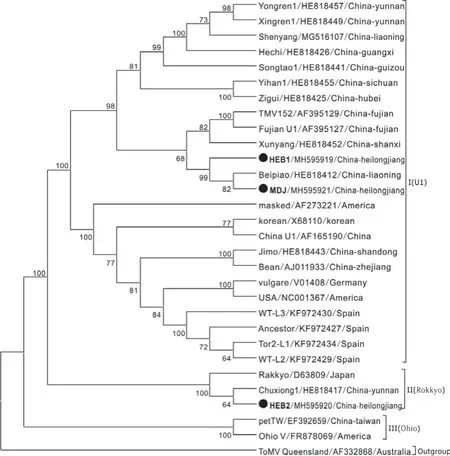
图3 基于HEB1、HEB2、MDJ和其他26个TMV分离物构建系统进化树Fig.3 Phylogenetic analysis of HEB1, HEB2, MDJ and other 26 TMV isolates
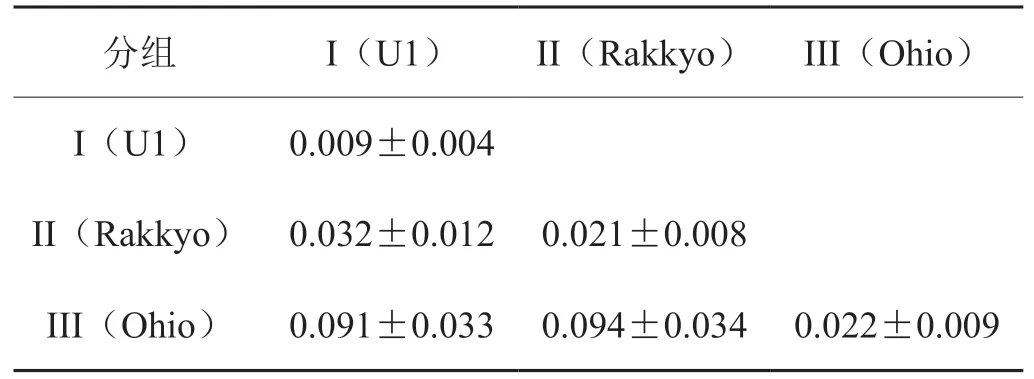
表6 组内和组间遗传距离分析Tab.6 Genetic distances within and between groups
2.5 选择压力分析
126 kDa、54 kDa、MP和CP四个蛋白dN/dS值均小于1(表7),表明TMV各蛋白处于负选择(或称为纯化选择)。其中,126 kDa蛋白的dN/dS值最大,而54 kDa的dN/dS值最小,说明126 kDa受到的选择压最小而54 kDa受到的纯化选择压最大。
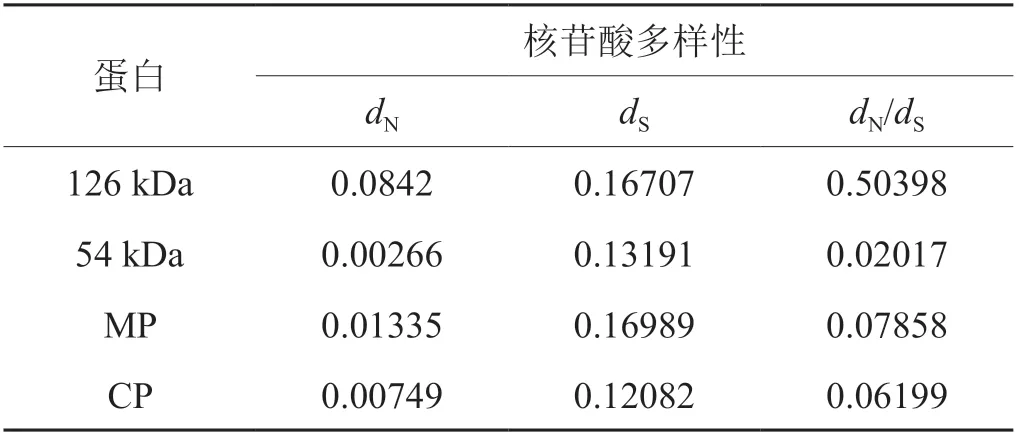
表7 TMV各蛋白选择压力分析Tab.7 Selection pressure analysis of TMV proteins

表3 HEB1、HEB2和MDJ与其他TMV分离物核苷酸的序列一致率Tab.3 Sequence identities between HEB1, HEB2 and MDJ with other TMV isolates at nucleotide level %
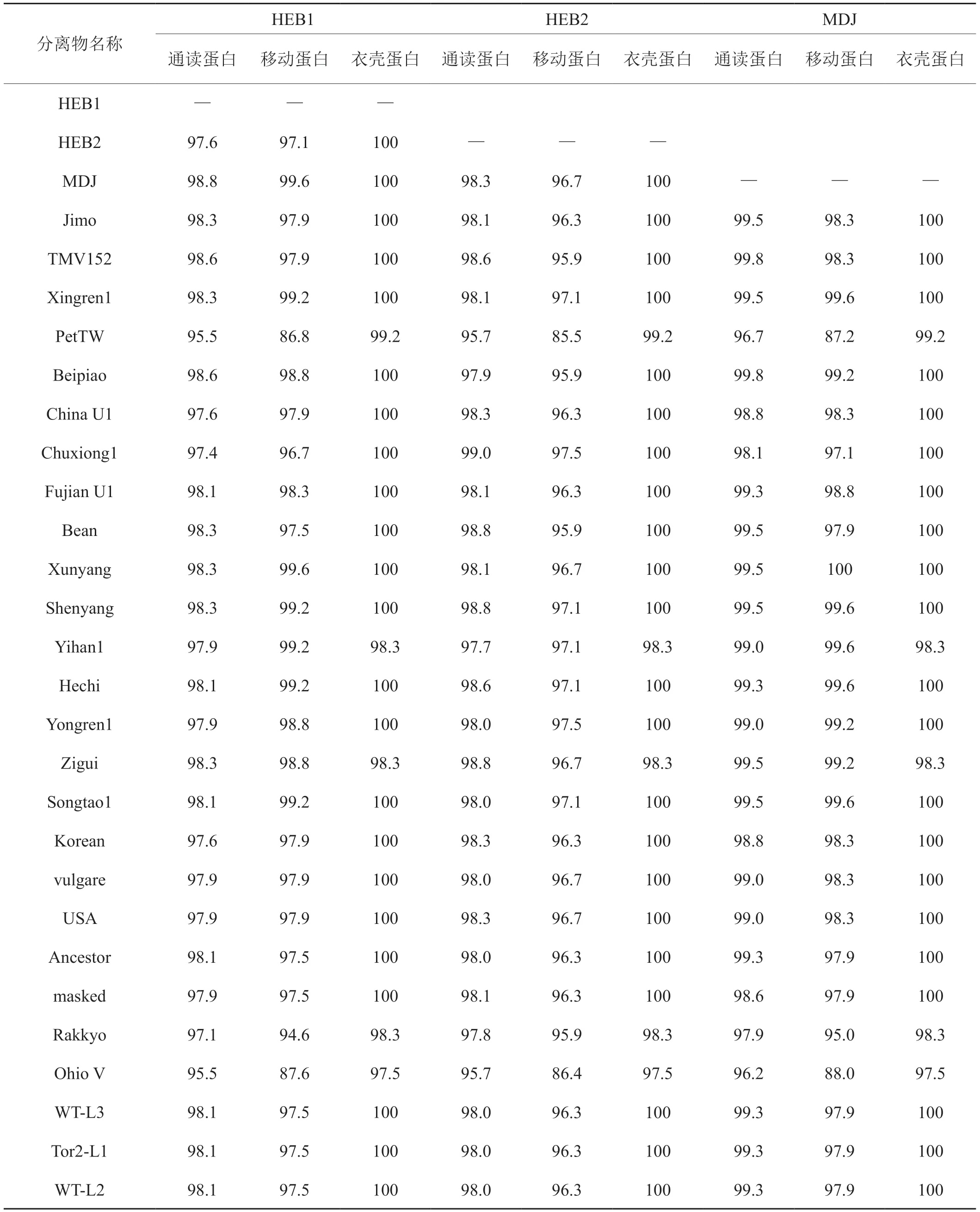
表4 HEB1、HEB2和MDJ与其他TMV分离物氨基酸的序列一致率Tab.4 Sequence identities between HEB1,HEB2 and MDJ with other TMV isolates at amino acid levels %
3 讨论
3.1 TMV株系划分及地理分布
以往的研究中TMV的株系划分主要依据生物学特性。谢联辉等[9]将福建烟草TMV划分为普通株系(TMV-C)、番茄株系(TMV-Tom)、黄色花叶株系(TMV-YM)和环斑株系(TMV-RS)4个株系,王劲波等[10]将山东烟区TMV划分为普通株系(TMVC)、坏死株系(TMVN)、黄化株系(TMVY)和环斑株系(TMVSR)4个株系。但TMV症状表型易受环境、寄主长势及侵染时间等条件影响[11]。近年来,TMV的鉴定和株系划分多依据核酸序列。Arguila等提出了划分十字花科烟草花叶病毒属病毒的标准:核苷酸序列一致率高于95%的为同一病毒,一致率85%~95%为密切相关的病毒。
Jakob等[12]对Ohio V株系进行全序列扩增并同其他Tobamovirus病毒比对4个ORF序列一致率,判定Ohio V与台湾矮天牛PetTW分离物属于同一株系。丁铭等[13]对云南的5个分离物扩增CP序列、秦西云[14]等对云南的38个分离物进行了CP序列扩增和分析,判定以上所有分离物均属U1株系。王莉爽等[15]对贵州的13个TMV烟草分离物进行CP扩增,发现云南分离物与贵州分离物CP序列一致率较高。刘天波等[16]根据地理相关性将TMV分为3个种群。将其系统进化树与图3比对,发现其3个种群基本对应I(U1)组的3个分支。刘金亮等[17]根据CP序列将TMV分为5个组。黄金光等[18]根据TMV的CP序列和症状特点将TMV分为2个株系。本文通过系统进化分析判断TMV分为3个组,其中分离物HEB1和MDJ属于U1株系,而且黑龙江烟区的U1株系与辽宁的Beipiao分离物聚类到一起,形成独立的一个分枝。HEB2属于Rakkyo株系,说明日本Rakkyo株系可能已经传播到了黑龙江。此前,该株系只在我国云南楚雄有发生。
3.2 TMV进化分析
选择压力分析表明TMV四个蛋白均处于负选择,说明寄主和环境等因素对TMV的选择压力很大,不利于蛋白变化,引起蛋白改变的突变体更容易被淘汰。与126 kDa蛋白和MP比较,CP面临的选择压力最大,这与蛋白氨基酸一致率的分析结果一致。在这4个蛋白中,54 kDa蛋白的dN/dS最小,说明该蛋白承受的选择压力更大,这可能与该蛋白是通过通读策略翻译产生的有关。
重组是病毒进化的重要途径之一,可改变病毒致病力[19],扩大寄主范围[20~22],有利于病毒适应当地环境。分离物HEB1存在重组信号,为MDJ与HEB2重组,说明Rakkyo株系可能与U1株系TMV分离物发生重组。
4 结论
本研究测定了黑龙江烟区3个TMV分离物的全基因组序列,进行了一致率、重组、系统进化和选择压分析,主要结论如下:
(1)HEB1、HEB2和MDJ的基因组全长均为6395核苷酸。HEB1和MDJ与辽宁分离物Beipiao一致率最高,分别为98.9%和99.6%;HEB2与云南分离物Chuxiong1一致率最高,为97.1%。
(2)HEB1是MDJ和HEB2的重组体。
(3)TMV根据全基因组序列分为3个组,其中HEB1和MDJ均属I(U1)组,HEB2属Ⅱ(Rakkyo)组。
(4)TMV的4个基因均处于负选择,其中衣壳蛋白基因的选择压力最大。
