Crohn’s-like acute severe colitis associated with Hermansky-Pudlak syndrome: A case report
2019-03-09PaulGirotCatherineLeBerreAstridDeMaissinMarieFreyssinetCarolineTrangPoissonArnaudBourreille
Paul Girot, Catherine Le Berre, Astrid De Maissin, Marie Freyssinet, Caroline Trang-Poisson, Arnaud Bourreille
Abstract BACKGROUND Hermansky-Pudlak syndrome (HPS) is a rare autosomal recessive disorder characterized by oculocutaneous albinism, platelet storage pool deficiency and systemic complications associated with ceroid deposition in the reticuloendothelial system. HPS types 1 and 4 are associated with Crohn’s disease(CD)-like gastrointestinal disorders, such as granulomatous enterocolitis or perianal disease. Cases of colitis can be particularly severe and, before the use of anti-tumor necrosis factor alpha (TNFα) therapy had become common, were reported as showing poor responsiveness to medical treatment.CASE SUMMARY We present the case of a 51-year-old albino woman who presented with acute severe colitis that led to the diagnosis of HPS. Histologic findings of biopsy samples showed chronic inflammation with deep ulcerations, and granulomas without caseous necrosis. Molecular genetic analysis confirmed HPS type 1, with a homozygous 27 base-pair deletion in exon 20 of the HPS1 gene. Once the patient’s bleeding diathesis was corrected by platelet transfusion, the granulomatous colitis responded dramatically to a medical treatment regimen that included corticosteroids, azathioprine and infliximab; this regimen is similar to that used in CD treatment. Although it remains unclear if the granulomatous enterocolitis in HPS is due to ceroid deposition or reflects the co-existence of CD and HPS, the fact that this case of HPS-related granulomatous colitis responded to the same therapeutic approach used in CD suggests that this type of colitis may result from HPS patients’ genetic susceptibility to CD.CONCLUSION We report a case of severe colitis that led to the diagnosis of HPS, which was responsive to azathioprine and infliximab.
Key words: Hermansky-Pudlak syndrome; Acute severe colitis; Infliximab; Azathioprine;Inflammatory bowel disease; Case report
INTRODUCTION
Hermansky-Pudlak syndrome (HPS) is a rare hereditary autosomal recessive disorder of lysosome-related organelles, characterized by oculocutaneous albinism and storage pool deficiency of platelets with increased bleeding tendency[1]. To date, there are eight subtypes, identified based on the particular genetic mutation responsible for the disorder[2]. Visceral disorders such as pulmonary fibrosis, colitis, cardiomyopathy or renal failure are associated with some but not all subtypes of HPS, with varying degrees of severity. Indeed, HPS types 1 and 4 have been reported as associated with inflammatory bowel disease (IBD)-like disorders of the gastrointestinal tract, such as granulomatous colitis, ileitis, enterocolitis, intestinal fistulization or perianal disease[3-5]. Their clinical, endoscopic and histologic features remain indistinguishable from Crohn's disease (CD) and ulcerative colitis, suggesting that their physiopathology might be related[3]. We report here a case of HPS type 1 that was diagnosed as a result of acute severe colitis.
CASE PRESENTATION
Chief complaints
A 51-year-old woman was admitted to the Gastroenterology Intensive Care Unit for hemorrhagic shock secondary to massive rectal bleeding.
History of present illness
The patient had lost about 15 kg over the past 5 mo, with recurrent episodes of diffuse abdominal pain that worsened following meals. Bloody diarrhea had started a few days before hospitalization, being intermittently mixed with stool first and progressing to massive rectal bleeding.
History of past illness, personal and family history
The patient had very limited medical history, comprised of very low visual acuity only. She reported having been abandoned at birth, and thus not knowing her family.
Physical examination upon admission
Initial physical examination demonstrated oculocutaneous albinism with whitish hair,pale skin, horizontal nystagmus and strabismus. Abdominal palpation elicited pain,and perineal examination did not show any anal fissure or fistula. The patient had a slight dyspnea, requiring low flow oxygen therapy, and pulmonary auscultation revealed bilateral crackles. She also had a severe undernutrition status, with a weight of 32 kg and a body mass index of 13.6 kg/m2.
Laboratory examinations
Laboratory findings were: hemoglobin of 5.4 g/dL (normal range: 13-18 g/dL), C-reactive protein (commonly known as CRP) of 73 mg/L (normal: < 5 mg/L), and hypoalbuminemia (1.6 g/dL; normal range: 3.4-5.4 g/dL). The white blood cell count(8.66 × 103/μL) and platelet count (268 × 103/μL) were normal. Bleeding time,activated partial thromboplastin time, and prothrombin time were normal. Stool samples cultures were negative, including for Clostridium difficile.
Imaging examinations
A computed tomography scan revealed diffuse bilateral interstitial infiltrates consistent with a severe pulmonary fibrosis (Figure 1A), and thickening of the descending/sigmoid colon associated with a transverse and ascending colon mild dilatation. Sigmoidoscopy showed active bleeding colitis, with continuous deep ulcerations (Figure 1B).
FINAL DIAGNOSIS
Histologic findings of biopsy samples showed chronic inflammation with deep ulcerations, extending into the muscularis mucosae, and granulomas without caseous necrosis. Molecular genetic analysis confirmed HPS type 1 with a homozygous 27 base-pair deletion in exon 20 of the HPS1 gene (c.2037_2064del).
TREATMENT
Intravenous methylprednisolone at a dose of 60 mg daily along with repeated transfusions of red blood cells concentrates was started upon admission. After 48 h,the patient’s CRP had decreased but the substantial rectal bleeding remained,necessitating additional transfusions. Thus, a colon rescue therapy was attempted with an infusion of infliximab at a dose of 5 mg/kg. Because of the HPS suspicion,platelet transfusions were also initiated, despite the normal findings for both platelet count and bleeding time; ultimately, this led to the bleeding ending.
Two weeks later, while the patient was getting better, the second infusion of infliximab was complicated by a severe anaphylactic reaction with bronchospasm that precluded continuance of this treatment. Azathioprine (50 mg daily) was started.
OUTCOME AND FOLLOW-UP
Two months later, deep remission was obtained, characterized by the absence of symptoms, normalization of inflammatory biologic markers, and mucosal healing(Figure 2). Unfortunately, the patient was not eligible for lung transplantation due to severe undernutrition and severity of pulmonary fibrosis, and she died of respiratory failure 3 mo later.
DISCUSSION
HPS was originally documented in 1959 by two Czechoslovakian physicians, who described two adults with a triad of albinism, hemorrhagic diathesis, and pigmented reticuloendothelial cells[1]. Except in the north-western quarter of the island of Puerto Rico, where HPS affects approximately 1/1800 persons and where approximately 1/22 persons are carriers of the gene, HPS remains extremely rare in the general population, with an estimated incidence between 1/500000 and 1/1000000[6]. HPS type 1 is the most common subtype and is associated with Puerto Rican heritage due to a founder mutation in this population. Diagnosis of HPS can be clinically suspected and is confirmed by molecular genetic analysis that allows classification into a particular HPS subtype (HPS 1-8).
Rarity of this syndrome can lead to delayed diagnosis and underlies the general lack of knowledge about its pathology, as was the case with our patient. HPS includes a platelet storage pool deficiency characterized by abnormally low contents of platelet α granules and/or δ granules[7]that results in a bleeding diathesis; this can be accompanied by normal findings in the usual blood tests, such as platelet count and bleeding time. There is no specific treatment, but transfusion of even limited numbers of normal platelets have been reported to alleviate the platelet dysfunction seen in HPS[7].
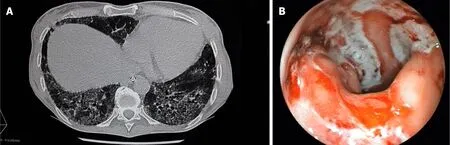
Figure 1 Computed tomography scan and sigmoidoscopy performed just after the patient’s admission to the Gastroenterology lntensive Care Unit. A:Chest computed tomography scan demonstrating an old and severe bilateral pulmonary fibrosis; B: Sigmoidoscopy showing active bleeding colitis with edema,erythema and linear deep ulcerations.
Granulomatous colitis was described as a complication of HPS for the first time in 1980[8]. Since then, many cases of inflammatory bowel disorders have been described,including those of colitis, enterocolitis or perianal disease[3,4,9-12]. These gastrointestinal complications are associated with HPS 1 and HPS 4 subtypes, occur in 20%-30% of the cases[3,4], and have been the cause of death in 9% of the deceased HPS patients in Puerto Rico[6]. The granulomatous colitis associated with HPS shares features with both ulcerative colitis and CD. In fact, clinical presentation and friable erythematous mucosa with ulcerations from rectum to cecum are suggestive of ulcerative colitis[10-12],but they present pathologic findings consistent with CD and can be associated with perianal and perirectal disease resembling the typical anorectal findings often seen in patients with CD[4,9,12].
Although HPS is caused by a single-gene defect that alters endosome trafficking and in spite of the fact that the proteins encoded by HPS1 and HPS4 genes are identified, the pathogenesis of the granulomatous colitis found in patients with HPS remains unclear. Moreover, it is also unclear if colitis associated with HPS is part of the syndrome, or if it represents an independent but associated process, such as CD[11].Initially, it was suggested that granulomatous colitis of HPS results from the accumulation of ceroid lipofuscin in intestinal macrophages because of the defects in synthesis, processing and trafficking of lysosome-related organelles. As there is no degradative pathway for ceroid, this accumulation was supposed to induce macrophages’ burst and a release of enzymes and cytokines, resulting in an inflammatory response[3,4,8,11]. But, some patients have had granulomatous colitis without ceroid pigments[10]and it has been noted that granulomas are not formed in relation to deposits of the ceroid-like pigments[8].
Thus, an alternative hypothesis has been proposed which is based on recent advances in the understanding of the pathophysiology of both HPS and CD[12]. HPS1 and HPS4 form a protein complex called the Biogenesis of Lysosome-related Organelles Complex 3 (BLOC-3), that triggers local activation of Rab GTP-ase proteins, called Rab32 and Rab38[13]. These small GTP-ases interact and co-locate with the protein LRRK2 to transport vesicles and recycling endosomes, and play an important role in the biogenesis and traffic of melanosomes and lysosomes. This system is disordered in HPS, accounting for the characteristic albinism[14], and LRRK2 has been recently identified by genome-wide association studies as a susceptibility gene for CD[15]. Therefore, this hypothesis suggests that granulomatous colitis in HPS results from genetic susceptibility to classical IBD.
In this case, medical treatment based on corticosteroids, azathioprine and one infusion of anti-tumor necrosis factor alpha (TNFα) therapy resulted in rapid improvement of clinical symptoms and endoscopic lesions. Among the nine published cases of HPS patients who received anti-TNFα therapy for a digestive complication[9-12], five had a good response, three necessitated surgical treatment, and the last one did not have any response to the anti-TNFα therapy but was improved by tacrolimus then vedolizumab[12]. The fact that therapeutic approaches similar to those for CD can also be effective in patients with HPS digestive complications reinforces the idea that HPS may be linked to CD. Further research is needed to determine the cause of the granulomatous enterocolitis of HPS, but all information gathered concerning the genetics of HPS enterocolitis might also shed light on CD pathogenesis.
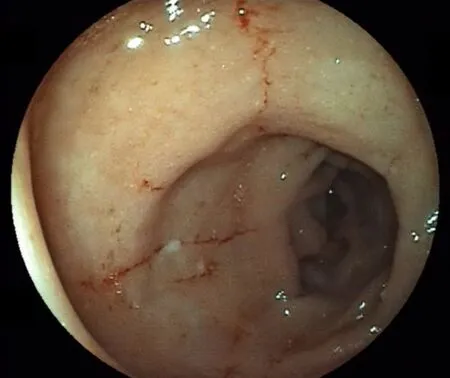
Figure 2 Sigmoidoscopy performed 2 mo after beginning the treatment. This image shows the improvement of edema and healing of linear ulcers.
CONCLUSION
We report herein the case of acute severe colitis in an albino woman, that led to the diagnosis of HPS. This granulomatous colitis responded dramatically to a therapeutic approach based on corticosteroids, azathioprine and infliximab, which is similar to the results seen with this regimen in IBD treatment.
杂志排行

World Journal of Gastroenterology的其它文章
- Current and future pharmacological therapies for managing cirrhosis and its complications
- Outcomes of per oral endoscopic pyloromyotomy in gastroparesis worldwide
- Dbx2 exhibits a tumor-promoting function in hepatocellular carcinoma cell lines via regulating Shh-Gli1 signaling
- Dynamic changes of key metabolites during liver fibrosis in rats
- Procyanidin B2 protects against diet-induced obesity and nonalcoholic fatty liver disease via the modulation of the gut microbiota in rabbits
- Triggers of histologically suspected drug-induced colitis