天然资源型化合物甾体皂苷元的化学研究
2019-03-08田伟生
田伟生
(中国科学院上海有机化学研究所,上海 200032)
面对人类社会发展面临的“资源日益匮乏”“环境污染加剧”问题和有机化学学科发展的历史和现状,20世纪90年代初作者提出了资源化学概念[1],并将其作为自己课题组的研究工作领域。资源粗放性利用是导致“资源浪费”“环境污染”的根本原因,没有资源浪费,就没有环境污染。为了实现物质资源在分子水平上的精准利用,化合物的化学研究成为化学家的挑战性任务。鉴于已知的化合物数目巨大(据2018年统计数字约14.4亿),本课题组仅仅选择了天然资源型化合物甾体皂苷元和非天然型化合物氟烷基磺酰氟两类化合物作为代表进行了系统研究。甾体皂苷元是一类含螺甾基本结构的化合物,在天然植物中它们则以与糖链接的皂苷形式存在。甾体皂苷元是甾体药物生产的基本生产原料之一。我国薯蓣皂苷元年生产量最高达4×106kg以上,剑麻皂苷元年产量约在5×105kg以上且大部分作为环境污染物被废弃。甾体皂苷元的生产历史超过半个世纪,其生产工艺十分成熟,因此甾体皂苷元的价格极为便宜,最低价格每公斤仅150多元人民币。此外,从薯蓣植物根茎中提取薯蓣皂苷元时仅利用了其资源的3%左右,其余的淀粉、纤维素和鼠李糖等尚未被利用,故其生产成本仍然存在降低空间。甾体皂苷元符合作为资源型化合物所定义的“规模大”、“价廉易得”要点。甾体皂苷元作为甾体药物生产基本原料并且有长达超过半个世纪的利用历史,但是由于人们对其化学性质研究了解的缺失从而导致甾体皂苷元利用过程中存在的严重环境污染问题长期得不到解决,甾体皂苷元利用过程中的资源浪费问题也没有引起人们关注,为此本课题组从20世纪90年代初开始展开了对甾体皂苷元化学及其应用的系统研究。本文简要地介绍了已经取得主要成果。
1 天然资源型化合物甾体皂苷元的反应及其应用
1.1 甾体皂苷元裂解物的洁净氧化降解反应
我国甾体药物生产基本上以薯蓣皂苷元作为起始原料,利用甾体皂苷元合成甾体药物首先需要氧化降解其为孕甾烯酮醇。如图1所示,实现这一转化的基本方法是依据美国化学家Marker研究给出的氧化降解反应[2]。Marker反应在20世纪40年代被应用于工业生产,我国从20世纪50年代起一直沿用至今。转化甾体皂苷元成为孕甾烯酮醇的关键反应是其裂解产物的氧化反应。Marker反应采用三氧化铬作为氧化试剂氧化降解甾体皂苷元裂解物,每氧化降解1×103kg甾体皂苷元将会产生约4×103kg含金属铬盐的废弃物。按照我国以往每年消耗4×106kg以上甾体皂苷元计算,将有约上千万公斤以上的含金属铬盐的废弃物产生。由于这些环境污染物分散在边缘山区的小化工厂中,它们的后续处理实际上无法进行。此外,在含金属铬废弃物中还含占降解产品近1/3(大约1×106kg)的手性分子被作为废弃物处理,造成甾体皂苷元资源利用过程中的严重浪费。
针对甾体皂苷元资源利用过程中的这一重大环境环境污染问题,本课题组系统地研究了用双氧水和氧气代替铬酐氧化降解甾体皂苷元的反应,研究发现用30%双氧水商品可以代替铬酐氧化降解甾体皂苷元裂解物成为相应的孕甾烯酮醇[3]。特别值得注意的是本课题组研发的反应不仅解决了甾体皂苷元铬酐氧化降解的金属铬环境污染问题,还可以方便地回收氧化降解产生的手性分子进一步转化成为宝贵的手性试剂,变废为宝,提高了甾体皂苷元的利用率,实现了甾体皂苷元资源“原子经济性利用”的目标。基于这一反应的甾体皂苷元洁净氧化降解技术在完成100 kg规模实验后,已经被转让给两家生产企业。毫无疑问,随着此技术的产业化推广应用,不仅将从源头解决我国甾体皂苷元资源利用过程中的资源浪费和环境污染的问题,也将促进我国甾体药物工业和手性药物的发展。


图1 薯蓣皂苷元氧化降解中Marker反应和本课题组研发反应的比较
1.2 双氧水作为氧化剂直接氧化降解甾体皂苷元的反应
在图1反应中,从甾体皂苷元制备孕甾烯酮醇时,首先需要通过高温、高压裂解反应转化甾体皂苷元成为相应的甾体皂苷元裂解产物。直接氧化降解甾体皂苷元的反应将有可能为工业部门提供更安全和便捷的甾体皂苷元资源利用技术,为此本课题组进一步研究了双氧水在各种酸性条件下氧化降解甾体皂苷元的反应(图2)。研究发现,30%双氧水在甲酸等介质中能以优异收率转化甾体皂苷元成为相应的Baeyer-Villiger氧化产物,即孕甾-16,20-醇类化合物和4-甲基戊内酯[4],还发现卤素(如溴和碘)能够改变反应的区域选择性给出相应的非正常的Baeyer-Villiger氧化产物,即孕甾-16,22-酸内酯类化合物和3-甲基丁内酯[5]。前一个反应已完成数十公斤规模试验,后一反应在试验室已反复进行了上百克规模的试验,它们均适合工业化生产应用。过氧三氟乙酸可直接氧化假甾体皂苷元,给出雄甾-16,17-醇类化合物。它们为高效利用甾体皂苷元资源提供了新的科学依据,为高效设计和合成甾体药物以及潜在应用价值的生物活性甾体分子提供了新机遇。

图2 用双氧水直接氧化降解甾体皂苷元的反应
1.3 甾体皂苷元双氧水直接氧化降解产物的反应
用双氧水代替铬酐氧化降解假甾体皂苷元得到的孕甾酮醇类化合物可以直接用于甾体药物合成,而甾体皂苷元双氧水直接氧化产物孕甾-16,20-醇和孕甾-16,22-酸内酯等在有机合成鲜有文献报道。为此,本课题组进一步研究了它们的反应(图3)。孕甾-3,16,20-三醇分子中3个仲羟基由于所处空间位置不同,它们对不同氧化剂表现出不同反应性能。与高价碘试剂,如Dess-Martin氧化试剂反应,可以选择性氧化其C16羟基[6]。孕甾三醇的微生物氧化与化学氧化具有互补性,其C20羟基更容易被微生物氧化得到16-羟基孕甾-3,20-二酮。孕甾三醇在溴化氢乙酸溶液(或乙酰溴)中反应以满意的收率给出C16溴代,C3和C20乙酰化产物,乙酰化溴代产物最好收率可达到89%[6-7]。溴代产物碎裂可得到C16(17)-甾烯化合物,16-溴代孕甾-3,20-二醇乙酸酯在碱性条件下可经Grob碎裂反应给出雄甾-16-烯化合物,它为甾体肌肉松弛剂和哺乳动物信息素合成提供方便[8-9]。
孕甾三醇乙酰化溴代产物还可被选择性地转化为含氧杂环丁烷结构单元孕甾分子[10]。该中间体在酸性条件下可选择性地在C20位置接受亲核试剂进攻,得到C20构型翻转的类天然甾体分子[11]。手性2,4-戊二醇曾经被用作手性辅助试剂合成手性分子,但是它在反应后被转化成为非手性化合物不饱和戊酮,故因使用成本过高未能得到应用。孕甾-3,16,20-三醇分子的16,20-二醇结构单元类似于手性2,4-戊二醇,可用其代替手性2,4-戊二醇作为手性合成辅助试剂,其反应所产生的孕烯酮醇化合物正好是甾体药物合成关键中间体。
孕甾-22-醛是合成具有胆固醇基本骨架的甾体生物活性分子的常用合成中间体,但其存在合成原料昂贵(如大豆甾醇或麦角甾醇)、自身化学稳定性差(容易异构化)、反应立体选择性低等问题。与此相反,孕甾-16,22-酸内酯合成原料便宜且制备方便,化学性质稳定且有优异的反应选择性,如图4所示。孕甾-22-醛与异戊炔基锂反应其立体选择仅1.5∶1,而孕甾内酯经加成/还原反应的立体选择性高达12∶1。这些恰好克服了利用孕甾-22-醛作为关键合成中间体合成天然胆甾类生物活性分子时存在的问题[12]。此外,由于孕甾内酯C16位存在羟基基团也为目标分子D环修饰带来方便。

图3 孕甾-3,16,20-三醇的反应

图4 甾体内酯加成/还原反应立体选择性与甾体-22-醛的比较
1.4 甾体皂苷元双氧水直接氧化降解产物在甾体药物和天然产物合成中应用
在专门研究资源型化合物甾体皂苷元的反应之前,合成甾体药物和天然甾体分子一般都采用甾体皂苷元的降解产物孕甾烯酮醇或去氢表雄酮作为关键合成中间体。双氧水直接氧化降解甾体皂苷元所得孕甾-16,20-醇和孕甾-16,22-酸内酯为甾体药物和生物活性天然甾体分子新合成策略和路线设计带来了机遇。研究表明用孕甾-16,20-醇和孕甾-16,22-酸内酯代替孕甾烯酮醇或去氢表雄酮作为关键合成中间体后确实显著提高了目标化合物的合成效率。
以猪外信息素合成为例(如图5所示),用孕甾三醇作为关键合成中间体,从薯蓣皂苷元出发经孕甾三醇的乙酰化溴代和碎裂等4步反应即可合成猪外信息素雄甾-16-烯-3-酮[8]。而文献以去氢表雄酮为关键合成中间体的方法,从薯蓣皂苷元出发,经Marker氧化降解反应和Beckemann降解反应等10步反应方可合成得到目标分子[13]。

图5 利用孕甾三醇合成猪外信息素的合成效率比较
吡嗪双甾体是一类化学结构新颖、抗肿瘤活性显著的海洋天然产物。本课题组以孕甾内酯作为关键中间体(如图6所示),从蕃麻皂苷元出发经19步反应,7.2%总收率的合成完成了其代表化合物cephalostatin 1北片断的合成。比文献报道以孕甾烯酮醇为关键合成中间体,从相同起始原料出发的35步反应,1.6%总收率的传统合成策略减少了16步反应(接近50%),并使总收率提高4.5倍[14]。

图6 利用孕甾内酯合成cephalostatin 1北片段的合成效率比较
以孕甾三醇和孕甾内酯等作为关键合成中间体,本课题组完成了苦楝甾醇(azedarachol)[15]、白前苷元C和D (glaucogenins C and D)[16-17]、17S-潘库溴铵(17S-pancuronium bromide)[18]、倍他米松(betamethasone)[19]、吡嗪双甾体cephalostatin 1[14]、油菜甾醇内酯(brassinolide)[20-22]、海洋甾醇clathsterol[23-25]、certonarsterol D2、D3和N1[20]和甾体生物碱clionamine D[26]等具有显著生物活性的天然甾体分子和甾体药物的高效合成(如图7所示)。分别从孕甾三醇和孕烯酮醇出发还完成了saundersiosides的形式合成[27]。
苦楝甾醇是从日本产苦楝根皮分离得到昆虫拒食剂,活性测试显示害昆虫AjrotissejetumDenis.的幼虫有拒食作用。本课题组以孕甾三醇为原料,经其C3-羟基消除、双羟化和C20-羟基反转并选择性酯化等12步反应,完成了苦楝甾醇的合成[15]。
白前苷元是一类双裂孕甾烷天然产物,其糖苷化合物普遍存在于白前、白薇、徐长卿以及马兰等中药中,生物活性研究表明该类化合物对α-正链RNA病毒有抑制活性,有可能开发成为抑制植物病毒的新农药。本课题组以孕甾三醇作为合成原料,经单线态氧烯反应和二价铁参与的烷氧基过氧化物碎裂两个关键反应,完成了双氢白前苷元C[16]和白前苷元D[17]的合成。
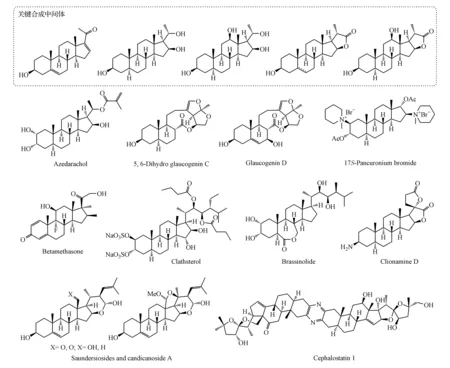
图7 利用孕甾三醇和孕甾内酯合成的部分甾体药物和天然产物分子
潘库溴铵类肌肉松弛剂已经被应用作为手术辅助药物,早期研究认为其17S-异构体有更好的活性,但是合成困难未能被选择开发。以孕甾三醇为原料,经溴代乙酰化反应、碎裂反应和消除等反应可以方便地完成了肌肉松弛剂17S-潘库溴铵的合成[18],为开发新型潘库溴铵类松弛剂药物提供了机会。
倍他米松是一种常用的糖皮质激素药物,从孕甾三醇出发,经溴代乙酰化、甲基铜锂试剂亲核取代、烯醇硅醚环氧化等反应完成了倍他米松关键中间体的高效合成[19],为糖皮质激素药物新合成路线设计与新技术进一步开发提供了科学依据。本课题组在合成倍他米松过程中还发现了甾体环氧烯丙醇的串联重排加成反应。
吡嗪双甾体cephalostatin 1是一类化学结构新颖的、具有显著抗肿瘤活性的海洋天然产物,也是至今化学结构最为复杂、在合成上最具有挑战性的天然产物之一,它的合成吸引了许多合成化学家的关注[28]。本课题组从蕃麻皂苷元降解得到的孕甾四醇和孕甾内酯出发,分别以27步反应,6.2%收率合成了其南片段,以19步反应,7.2%收率合成了其北片段,然后经4步反应进行南/北片段连接完成了cephalostatin 1的合成[14],提供了一个高效实用的合成此类天然产物的方法。本课题组还从蕃麻皂苷元降解内酯出发合成了另一类结构独特吡嗪双甾体ritterazine N的类似物[29]。
油菜甾醇内酯是首先被从油菜花粉中分离得到甾体植物生长激素,后发现它及其类似物广泛存在于植物体内。由于其对农作物的增产作用和其从自然界直接获取的困难性(在干燥油菜花粉中含量约百万分之一),油菜甾醇内酯类化合物合成一直受到合成化学家的关注。本课题组从薯蓣皂苷元降解得到的甾体内酯出发,经20步反应,以7%总收率完成油菜甾醇内酯的克级合成,为油菜甾醇内酯类植物生长激素的基础研究提供了必须的样品[21-22]。
海洋甾醇clathsterol是从红海海绵Clathria中分离得到的一种甾醇硫酸盐,对HIV-1逆转录酶具有很好的抑制活性。为了确定其侧链的立体化学并发展高效合成方法,本课题组从剑麻皂苷元氧化降解所得孕甾内酯出发完成了C22,C23-不同构型的clathsterol异构体的合成,通过波谱数据比较确定其立体化学为22S、23S构型。并以21步反应,11%总收率完成clathsterol硫酸盐的合成[25]。
自非洲海域的海绵体分离出的甾体生物碱clionamine D,具有罕见的γ-螺双内酯结构,并且具有良好的细胞自噬调节作用。从剑麻皂苷元氧化降解产物孕甾内酯出发、经Schenck烯反应、以及三价锰介导的[3+2]环加成等10步反应,本课题组以51%总收率完成它的合成[26]。通过初步生物活性研究还发现它的3-位异构体和3-羟基和羰基类似物具有相似生物活性,初步判断clionamine D的生物活性与其γ-螺双内酯结构有关。
孕甾内酯作为天然甾醇合成关键中间体的策略也被姜标等[30]用于海洋天然甾醇Certonarsterol D2的合成;Morzycki等[31]用孕甾内酯作为合成F环为糖结构单元的螺甾分子“glycospirostanes”的原料;俞飚等[32]以孕甾内酯作为关键合成中间体完成了海星多羟基甾体皂苷Linckoside A和B的全合成(如图8所示)。显而易见,孕甾三醇和孕甾内酯等作为关键合成中间体在天然产物合成应用研究中目前仅仅是一个良好的开端,随着时间推移它们在生物活性的甾体及相关分子合成中会发挥越来越重要的作用。

图8 海洋甾醇Certonarsterol D2和Linckoside A和B苷元的合成
1.5 甾体皂苷元氧化降解所获手性试剂及其在有机合成中的应用
如上所述,基于双氧水氧化甾体皂苷元反应而发展的甾体皂苷元洁净氧化降解技术已经开始向生产企业转让。这些技术在生产企业全部推广实施后,每年将有可能生产出近百万公斤带手性甲基侧链的手性试剂和手性合成原料,如何使这些从废弃物中获得的宝贵手性试剂和原料应用于工业生产、服务于社会是研究工作的追求目标之一。
尽管文献中已经报道了许多合成带甲基侧链的手性分子的方法,然而已知的手性试剂中,带甲基侧链的手性试剂品种少,价格昂贵。如Aldrich试剂公司销售的最常见的2-甲基-3-羟基丙酸甲酯,每克售价450元,作为工业原料每公斤也需要6万元(甾体皂苷元氧化降解主产品孕烯酮醇每公斤也仅仅800元左右)。由此可见我国在甾体皂苷元资源利用过程中不仅存在严重环境污染,同时存在巨大资源浪费。若按照2-甲基-3-羟基丙酸甲酯工业原料价格计算,我国每年在甾体皂苷元资源利用过程中废弃手性试剂价值约在上百亿人民币。在从甾体皂苷元氧化降解废弃物中回收到手性化合物4R-甲基-5-羟基戊酸内酯和4R-甲基-2,5-二羟基戊酸-δ-内酯后,本课题组系统地研究了它们的反应,将它们转化成为一些便于保存和方便使用的手性试剂,图9列举部分手性试剂的化学结构[33-34]。这些手性试剂和原料为手性药物、农药和昂贵的香精香料等的高效合成提供了物质基础。
传统农药已经在防治病虫害促进农作物丰产方面发挥了巨大的作用,但是它们对土壤和水资源的造成的严重污染也越来越受到人们关注,发展高效的、无环境污染的新农药是社会对化学家提出的挑战性任务。昆虫信息素是存在于昆虫体内的内源性激素物质,能够调控昆虫群体的各种行为。昆虫信息素应该是十分有效的防治各种虫害的药剂,但由于其在昆虫体内含量微少导致从天然来源获取它们十分困难。与此同时,由于合成所需的手性原料和试剂品种少、价格昂贵也限制这些昆虫信息素的高效合成。为了解决传统农药带来的环境污染问题,发展高效的合成各种不同昆虫的信息素的方法,本课题组利用上述已经发展手性试剂完成了一些光学活性的昆虫信息素,如松叶蜂信息素[35-36]、玉米根虫信息素[37]、昆虫保幼激素[38-39]、黄瓜甲虫信息素[40]、蚂蚁信息素[41]、谷盗聚集信息素tribolure[42]等(如图10所示)。

图9 由戊酸-δ-内酯衍生的部分手性试剂

图10 由戊酸-δ-内酯衍生的手性试剂合成的药物和天然产物
松叶蜂广泛分布于欧洲、亚洲以及北美洲,是危害针叶林的主要害虫,它的合成受到许多合成化学家的关注[37]。其中,Mori等[43]从光学活性香茅醇合成松叶蜂时需经过12步反应合成才能得到关键中间体4-甲基-溴代十二烷,而利用从甾体皂苷元氧化降解所获得的手性试剂5-溴代-4-甲基戊酸甲酯,只需3步反应就可合成(如图11所示)。

图11 松叶蜂性信息素合成方法效率比较
昆虫保幼激素Hydroprene是美国Zoecon公司开发的一种有效的具有保幼激素活性的昆虫生长调节剂,该药剂对烟芽夜蛾、埃及伊蚊、大蜡螟、黄粉甲、家蝇、蟑螂等均有良好的防治效果。该药剂还是美国目前唯一一种经登记许可在厨房、食品储藏室、医院和一些敏感部位使用的昆虫生长调节剂。利用4R-甲基戊内酯衍生的手性试剂为原料,本课题组完成了昆虫保幼激素hydroprene以及methoprene的合成[37-39]。
许多香精香料的成分都含有手性甲基侧链,如香瓜醛、香茅醛、异胡薄荷醇、薄荷呋喃、麝香酮、Citralis Nitrile、Tropional、Firsantol、Rose oxide、Cedrol等。本课题组利用从甾体皂苷元氧化降解所获得的手性试剂完成了麝香酮[44]、薄荷呋喃[45]、香瓜醛[45]、香茅醛[45]和Citralis Nitrile[46]等香料手性分子合成。
本课题组也利用从甾体皂苷元氧化降解所获得的手性试剂于药物和具有生物活性的天然产物合成。如维生素E(vitamin E)[47-49]、抗心衰药物Entrestro的活性组分之一沙库必曲(sacubitril)[50]、海洋天然产物didemnaketal A的C1-C8核心片段[51]和C9-C23[52]等高效合成。
从甾体皂苷元氧化降解废弃物中获取的手性试剂也被其他研究团队用于天然产物合成。马大为等[53]在halipeptins A-D海洋环肽合成中利用了4R-甲基戊内酯。涂永强等[54]在合成海洋天然产物didemnakatol A中向本课题组实验室购买了相应的手性试剂。
手性甲基侧链的结构单元广泛存在于生物活性天然产物、医药、农药和香料分子结构中,以往由于制备困难限制这些分子的合成与应用。随着甾体皂苷元洁净氧化反应在工业生产的推广应用,手性甲基侧链的结构单元的试剂和原料将能够实现大规模生产,届时价廉的且多样化手性试剂和原料的提供必然促进手性药物、农药和精细化工产品的发展。
1.6 甾体皂苷元E/F的开环反应及其在甾体天然产物分子合成中应用
胆固醇是存在于人类、哺乳动物、昆虫等动物体内最基本甾体化合物,它们是动物体内细胞膜的组织部分,也是各种甾体激素物质的合成前体。植物体内的植物甾醇化学结构类似于胆固醇,真菌体内的麦角甾醇化学结构也胆固醇十分相近。已知天然界存在上千种具有胆固醇基本骨架的生物活性分子,它们是生物体内的重要活性物质,也是发展新医药、新农药的先导分子。甾体皂苷元的E/F螺环开环产物不仅具有胆固醇相同的基本骨架,而且其已经存在的官能团为合成具有胆固醇基本骨架的各类天然产物分子提供了方便(如图12所示)。利用甾体皂苷元完整骨架合成具有胆固醇基本骨架的各类天然产物分子的关键是需要了解甾体皂苷元的E/F螺环的反应性能。

图12 胆固醇、天然产物OSW-1苷元与甾体皂苷元结构比较
有机反应的普适性研究近年来在我国有机化学界受到越来越多的关注,这是因为所有官能团或结构单元的转化反应都受到反应底物的严格限制。反应应用范围严格受制于反应底物,许多常见反应不适合于资源化合物。为了精准利用资源型化合物,故它们的反应及其应用研究必须给予重视。
甾体皂苷元E/F螺环为缩酮官能团,已知缩酮很容易酸水解成为相应链状酮醇化合物,然而在研究中发现甾体皂苷元E/F螺环缩酮在酸水解条件下根本无法得到相应的链状酮醇化合物。设想通过保护缩酮开环后的羰基或羟基促进其开环反应,为此本课题组研究了甾体皂苷元的开环缩硫酮化和开环乙酰化反应。在研究甾体皂苷元的开环硫缩酮化反应中发现在路易斯酸存在下单硫醇与甾体皂苷元反应给出F环开环的26-硫代产物;当与双硫醇反应时则给出F环开环、分子内氧化还原以及硫缩醛化的26-硫缩醛产物;当与硫化氢反应时直接给出了硫代甾体皂苷元[55-56]。这一反应为合成硫代甾体皂苷提供了有效的方法,它也被应用于利用薯蓣皂苷元完整骨架合成具有显著抗肿瘤生物活性的天然产物OSW-1的苷元[57-58]。16-羟基甾体皂苷元在相同反应条件下与硫醇反应给出相应的16-单或双硫缩酮产物[59-60],它们经乙酰化反应得到相应的16-烯基硫醚产物。这些反应已被应用于名贵中药重楼活性成分苷元Pennogenin的合成[61]。此外,16-硫缩酮基团存在提高了胆甾-22-酮还原的立体选择性,通常给出C22S-构型还原产物。
路易斯酸催化的甾体皂苷元溴代(及卤代)反应随甾体皂苷元结构不同而给出不同结果。16-羟基甾体皂苷元给出的是26-溴代E/F螺环开环产物,5,6-二溴代-16-羟基薯蓣皂苷元溴代给出相应的26-溴代E/F螺环开环产物用锌粉脱溴时可直接给出16,22-二氧代胆固醇,也可选择性脱溴给出16,22-二氧代-26-溴代胆固醇[60]。前者应用于合成OSW-1苷元[62],后者应用于甾体生物碱的合成[63]。甾体皂苷元直接溴代给出的主要产物是26-溴代E/F螺环开环产物[64-65],它为C26为氮取代的甾体生物碱苷元合成提供了方便[66]。12-氧代甾体皂苷元(如蕃麻皂苷元)反应则可以得到16,26-二溴代产物[64],这为从甾体皂苷元完整骨架合成天然产物jaborosalactones类化合物提供了方法[67]。在甾体皂苷元26-硫代、卤代开环反应的基础上,本课题组考察了其26-胺化开环反应。研究发现在路易斯酸存在下,磺酰胺与甾体皂苷元反应可以获得胺化开环反应结果,此反应也被应用于甾体生物碱苷元,如中药龙葵生物碱合成[68]。
为了实现甾体皂苷元E/F螺环开环,本课题组早在20世纪90年代就研究了其乙酰化开环反应[69]。由于反应时间未能很好控制,得到了E环开环后进一步乙酰化反应产物[70],此反应在随后被Fernabdez-Herrere等[71-72]通过控制反应时间在15 min后得以实现。这一反应特别适合呋咱型甾体皂苷的高效合成,但是仍然不适合其D环结构修饰,原因是C16-乙酰基脱去后即刻环化回到甾体皂苷元E/F螺环。为此,本课题组进一步研究了甾体皂苷元的乙酰化开环/还原或腈基化反应[64],反应产物适合于进一步进行其D环结构修饰。
甾体皂苷元F环可以方便通过还原打开,利用26-酰基正碳离子的分子内活化作用可以实现甾体皂苷元E环的开环[73],但是反应伴随的重排反应的发生而导致收率显著下降。对此反应本课题组通过添加卤代试剂抑制了重排反应给出一个新的溴代分子内酰化开环反应[74]。此方法操作方便、反应收率高,适合于C25-位为R或S两种构型立体化学的天然产物合成。这一反应被成功地应用于天然甜味剂Osladin苷元[75]、中药重楼活性成分皂苷元Pennogenin[64]、中药小百部活性成分Aspafiliosides E和F的苷元[76-77]、天然抗菌活性甾体Boophiline以及甾体生物碱solasodine、solanidine、demissidine[75,78]等的高效合成以及线虫生长调节剂Dafachronic Acid A的形式合成(如图13所示)。
海洋天然产物Saundersiosides是一类具有24(23→22)-移-胆甾烷结构和显著的抗肿瘤活性的胆甾烷分子。最近本课题组研究发现甾体皂苷元乙酰化开环产物经二醋酸碘苯引发的Favorskii重排可以高效转化成为天然产物Saundersiosides的基本骨架(如图14所示)。这一反应为甾体皂苷元资源利用和Saundersiosides等海洋天然产物高效合成进一步提供了基础[79]。
2 展 望
针对甾体皂苷元资源利用过程中存在的“资源浪费”和“环物污染”问题,本课题组系统地研究了甾体皂苷元的化学,在其反应和应用方面取得了系列阶段性成果,给出了原子经济性利用甾体皂苷元化合物资源的策略和方法。对于已经实现了按照原子经济性利用的资源型化合物的高效利用策略,将会在非天然资源型化合物氟烷基磺酰氟的化学中介绍。用30%双氧水代替传统方法中用铬酐氧化降解伪甾体皂苷元成为孕烯酮醇的技术虽然已经转让相关企业,但是真正实现产业化还需要企业界的同仁们继续努力。本课题组发展的直接氧化降解甾体皂苷元成为相应的孕甾三醇和甾体-22-酸内酯的技术避免了使用伪甾体皂苷元降解所需要的剧烈裂解反应,反应条件更加温和、安全、高效,但能否为企业界所接受同样需要时间。仅仅把甾体皂苷元作为甾体药物合成原料的传统思维和甾体皂苷元化学研究的迟缓导致了其利用过程的“资源浪费”和“环境污染”问题。从资源化学的理念考虑,甾体皂苷元不仅是甾体药物生产的基本原料,同样是含手性甲基侧链的药物或功能有机化合物的合成原料。从甾体皂苷元洁净氧化降解所获得的手性试剂重量虽然只占所获甾体药物合成关键中间体孕烯酮醇的二分之一,但其每公斤单价确比孕烯酮醇高出数十倍。毫无疑问,如何充分利用从甾体皂苷元洁净氧化降解所获得的手性试剂是提高甾体皂苷元资源利用价值的关键。在分子水平上精准利用资源是解决人类社会面临的“资源日益匮乏”和“环境污染加剧”问题的根本策略,而资源型化合物的化学研究无疑是为精准利用资源提供科学依据和实用技术的必经之路。资源型化合物的化学(资源化学)研究是社会发展的需求,是有机化学及其相关科学学科发展的需要。资源化学研究也新药发展密不可分,已知在同类化合物中其生物活性强度与存在规模成反比,如何将规模大的资源型化合物高效转化成为药物或高生物活性的分子必须依赖于对资源型化合物化学知识的了解。作者希望借课题组在资源化学领域里取得的阶段性研究成果之“砖”引起更多学者对资源化学的关注和投入,产生更多、更好的资源化学研究成果造福人类社会。
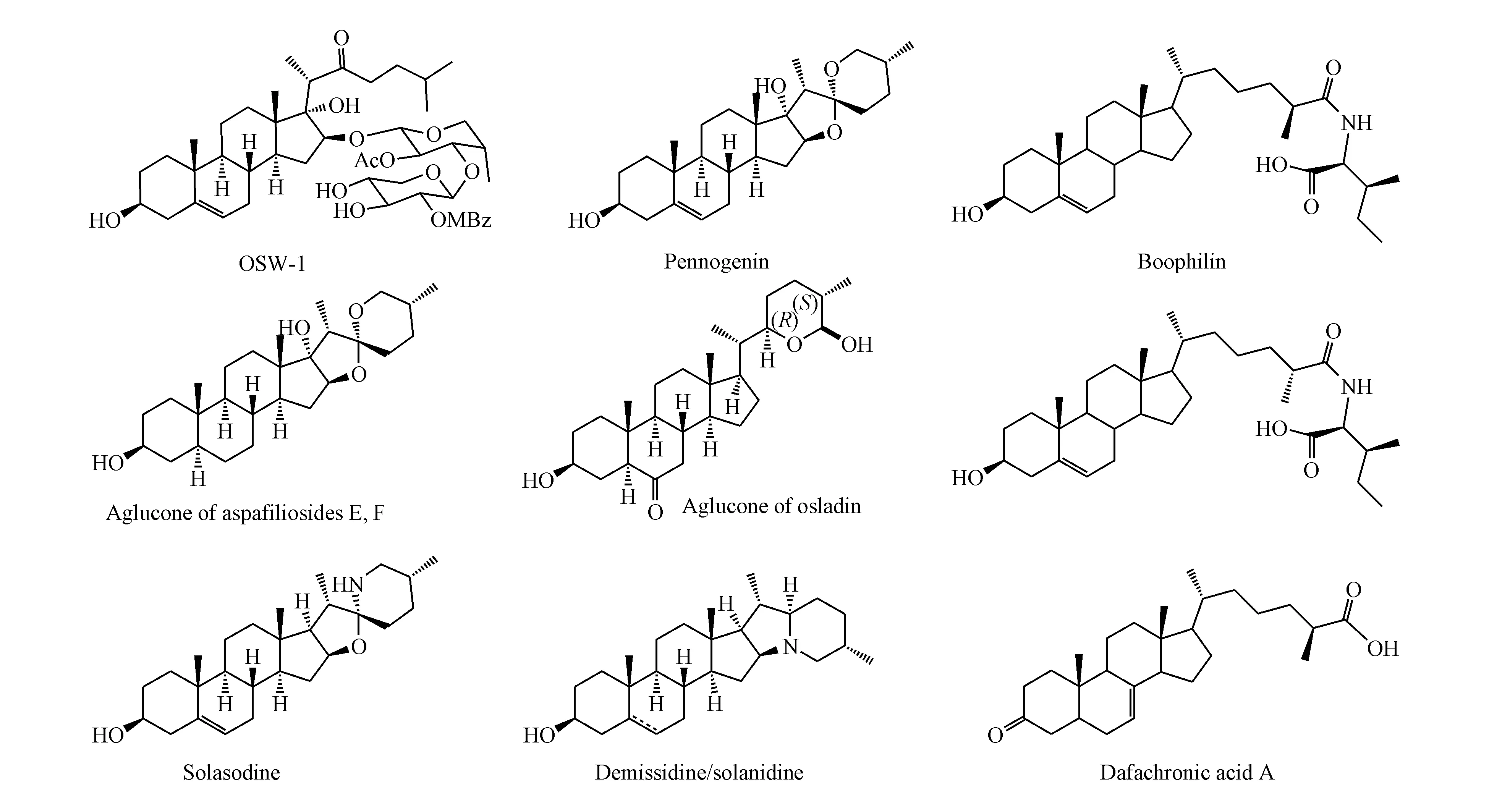
图13 利用甾体皂苷元完整骨架合成的部分天然甾体分子
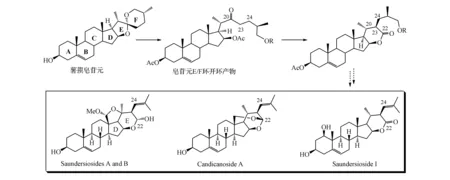
图14 Saundersioside基本骨架合成
致谢:随着中国社会改革开放,作者在1978年有幸作为廖清江教授的研究生进入中国药科大学(原南京药学院)学习并走上了化学研究之路。在中国改革开放四十周年之际,作者应母校《中国药科大学学报》编辑部及尤启冬教授的邀请,将本课题组的部分研究工作总结,以回报母校培养之恩。本文是基于作者为中国科协撰写的2015- 2017年化学学科进展报告中《资源化学进展》专题报告改写而成。在此还要特别感谢中国药科大学向华教授为本文改写付出辛勤劳动。
