Improvements of TKC Technology Accelerate Isolation of Transgene-Free CRISPR/Cas9-Edited Rice Plants
2019-03-06HeYubingZhuMinWangLihaoWuJunhuaWangQiaoyanWangRongchenZhaoYunde
He Yubing, Zhu Min, Wang Lihao, Wu Junhua, Wang Qiaoyan, Wang Rongchen, Zhao Yunde, 4
Improvements of TKC Technology Accelerate Isolation of Transgene-Free CRISPR/Cas9-Edited Rice Plants
He Yubing1, 2, 3, Zhu Min1, 2, Wang Lihao1, 2, Wu Junhua1, 2, Wang Qiaoyan1, 2, Wang Rongchen1, Zhao Yunde1, 4
(National Key Laboratory of Crop Genetic Improvement and National Center of Plant Gene Research (Wuhan), Huazhong Agricultural University, Wuhan 430070, China; College of Life Science and Technology, Huazhong Agricultural University, Wuhan 430070, China; College of Plant Science & Technology, Huazhong Agricultural University, Wuhan 430070, China; Section of Cell and Developmental Biology, University of California San Diego, La Jolla, California 92093-0116, USA)
Elimination of the CRISPR/Cas9 constructs in edited plants is a prerequisite for assessing genetic stability, conducting phenotypic characterization, and applying for commercialization of the plants. However, removal of the CRISPR/Cas9 transgenes by genetic segregation and by backcross is laborious and time consuming. We previously reported the development of the transgene killer CRISPR (TKC) technology that uses a pair of suicide genes to trigger self-elimination of the transgenes without compromising gene editing efficiency. The TKC technology enables isolation of transgene-free CRISPR-edited plants within a single generation, greatly accelerating crop improvements. Here, we presented two new TKC vectors that show great efficiency in both editing the target gene and in undergoing self-elimination of the transgenes. The new vectors replaced the CaMV35S promoter used in our previous TKC vector with two rice promoters to drive one of the suicide genes, providing advantages over our previous TKC vector under certain conditions. The vectors reported here offered more options and flexibility to conduct gene editing experiments in rice.
genome editing; suicide gene; transgene killer CRISPR; Cas9; transgene-free
CRISPR gene editing technology enables efficient targeted modifications of genes/DNA elements in virtually any transformable organisms, providing unprecedented tools for agricultural improvements. Ever since the first reported CRISPR/Cas9-mediated editing events in eukaryotes (Cong et al, 2013; Mali et al, 2013), CRISPR technologies have been steadily improved in several fronts: 1) The types of modifications that CRISPR can introduce have been remarkably expanded. Initially, CRISPR/Cas9 is able to produce small insertions/ deletions when CRISPR/Cas9 inducing double stranded DNA breaks (DSBs) are repaired by the error prone non-homologous end-joining (NHEJ) pathway (Jinek et al, 2012). Scientists now can use CRISPR to generate targeted point mutations including both A∙T to G∙C (Gaudelli et al, 2017; Hua et al, 2018; Yan et al, 2018) and G∙C to A∙T conversions (Komor et al, 2016; Li J Y et al, 2017; Lu and Zhu, 2017; Ren et al, 2018), and to introduce new functions through homology-directed DNA repair (HDR) (Cermak et al, 2015; Chu et al, 2015; Sun et al, 2016; Wang et al, 2017; Li J Y et al, 2018; Li S Y et al, 2018a). 2) Editable target DNA sequences have been expanded enormously. All CRISPR nucleases require a protospacer adjacent motif (PAM) sequence (Kleinstiver et al, 2015). The Cas9 nuclease requires the target sequence adjacent to the 5′-end of the NGG PAM sequence (Jinek et al, 2012). Identification of other nucleases such as Cpf1 that have a different PAM requirement allows using CRISPR to edit different target sequences (Zetsche et al, 2015; Fonfara et al, 2016; Zetsche et al, 2016). Newly developed Cas9 variants and Cpf1 variants further widen the choices of target sequences (Kleinstiver et al, 2015; Burstein et al, 2017; Gao et al, 2017; Murovec et al, 2017; Hu et al, 2018; Li S Y et al, 2018b; Zhong et al, 2018). 3) The fidelity of nucleases has been improved to limit off-target effects (Casini et al, 2018; Hu et al, 2018). 4) Nuclease-dead enzymes such as dCas9 and dCpf1 have been repurposed for applications other than gene editing. Fusions of a functional domain or an enzymatic activity with dCas9 enable transcriptional activation/repression (Gilbert et al, 2013; Konermann et al, 2015; Li Z X et al, 2017; Tak et al, 2017), epigenetic modification (Vojta et al, 2016; Liu et al, 2016; Huang et al, 2017; Stepper et al, 2017; Gallego-Bartolome et al, 2018), and visualization of genetic elements such as telomeres (Tanenbaum et al, 2014; Dreissig et al, 2017; Zhou et al, 2017). At present, scientists have the ability to conduct virtually any DNA modifications in living cells, revolutionizing both medical research and agricultural improvement, but there are still major challenges in using gene editing in both therapeutics of diseases and improvements of crops.
There are unique challenges in using CRISPR gene editing technology for crop improvements compared to the medical field. One of the concerns is that CRISPR/Cas9 constructs may escape to the environment through the spread of pollen particles. Another concern is that the existence of transgenes such asin crops invokes food safety worries. Therefore, it is critical to remove the CRISPR constructs after the target gene is edited as designed. Interestingly, it seems that biomedical field hardly has any concerns for the removal of CRISPR constructs. We have been keen on developing technologies that efficiently and quickly removeand other transgenes after the gene editing task is completed in plants for the aforementioned reasons. In, we tethered the CRISPR/Cas9 editing unit with a mCherry unit, which generates seeds with bright red fluorescence (Gao et al, 2016; He et al, 2017a). Our fluorescence protein-based approach enables efficient and reliable isolation of Cas9-free and editedplants. In rice, we developed the transgene killer CRISPR (TKC) technology (He et al, 2018) to enable self-elimination of the transgenes after target gene is edited. The TKC technology links the CRISPR/Cas9 gene editing cassette with two suicide gene cassettes that kill transgene-containing pollen and embryos. The key to the success of TKC technology is to temporally separate the expression ofand the suicide genes. The gene editing components including Cas9 and gRNAs are expressed during transformation, callus stage, and vegetative growth phase. Upon transition to the reproductive phase, the suicide genes are expressed to kill all transgene-containing pollen and embryos. Therefore, only transgene-free seeds are produced, enabling the isolation of gene-edited transgene-free rice plants in just a single generation.
Our first generation TKC technology employsandas the suicide genes (He et al, 2018). We placed thegene under the control of a CaMV35S promoter. We expressedusing the REG2promoter, which is active during embryo development. The CaMV35S-CMS2 and REG2-BARNASE cassettes lead to successful self-elimination of transgenes without affecting editing efficiency. Despite the apparent success of our initial TKC technology, we believe that it can be further improved for several reasons. Firstly, CaMV35S promoter is relatively weak in monocots than in dicots (McElroy et al, 1990). Previous studies have shown that CaMV35S promoter is especially weak in floral organs, particularly in the microspore cells (Wilkinson et al, 1997). It is also reported that transgene silencing occurs when the CaMV35S promoter is used to overexpress a target gene (Hall et al, 2001). The CaMV35S promoter contains a region that is easily methylated, which leads to a decrease in the promoter activities (Weinhold et al, 2013). Secondly, CMS2is a rice endogenous mitochondrial toxic protein (Wang et al, 2006). Although the presence of CMS2 does not cause lethality of somatic cells except pollen cells, previous studies have shown that accumulation of CMS2 protein causes an increase in mitochondrial superoxide levels, which makes plants more sensitive to drought than those without CMS2 (Yu et al, 2015).
In this study, we improved our TKC technology in rice by replacing the CaMV35S promoter used in our previous TKC plasmids with two different rice promoters. We used the rice ACTIN1 promoter, which is strong and constitutive (McElroy et al, 1990), to analyze the impact of a broadly expressed CMS2 on our TKC technology. We also used a pollen-specific promoter OsGEX2 promoter (Cook and Thilmony, 2012) to drive CMS2 expression in pollen cells. Both strategies led successful isolation of transgene-free and edited rice plants within a single generation.
Materials and Methods
Rice materials
Rice cultivar Zhonghua 11 [L. ssp. geng ()] was used for the study. Rice plants were grown in normal fields under natural conditions in Wuhan City, Hubei Province, China.
Construction of the TKC plasmids
We prepared several DNA units for constructing the TKC plasmids. In TKC1.1, the OsACTIN1 promoter was derived from the plasmid pActin1:RGR1-RGR2 (He et al, 2017b). In TKC1.2, the OsGEX2promoter was amplified from the genomic DNA of Zhonghua 11 using the primers OsGEX2P- TAF and OsGEX2P-TAR, and then cloned into the T-vector pEASY-T5 (TransGen Biotech, Beijing, China) following the manufacturer’s instructions to construct the plasmid pOsGEX2P-TA. The REG2-BARNASEexpression cassette was present in the TKC1.0 plasmid (He et al, 2018). The primers used in this study are listed in Supplemental Table 1.
The TKC plasmids were constructed using the CRISPR vector pCXR9Tas the backbone, which contains the UBIQUITIN promoter-Cas9cassette and the RBCS-E9 terminator (He et al, 2018). The other cassettes were amplified by PCR and cloned into pCXR9T using Gibson assembly (Gibson et al, 2009). In TKC1.1, we used plasmids pActin1:RGR1-RGR2and pCMS2-TA (He et al, 2018) as templates to construct the Actin1p-CMS2 expression unit by overlapping PCR reactions with the primer pairs Actin1P-F/Actin1P-R and CMS2-TAF/CMS2-R, respectively. The PCR product was inserted into theI site in pCXR9T by Gibson assembly to produce the plasmid Actin1P-CMS2- pCXR9T. In TKC1.2, the OsGEX2P-CMS2 expression unit was generated by overlapping PCR reactions with pOsGEX2P- TA and pCMS2-TA as templates and OsGEX2P-F/OsGEX2P-R and CMS2-TAF/CMS2-R as primer pairs. The OsGEX2P- CMS2 unit was cloned into theI site in pCXR9T. The resulting plasmid was called OsGEX2P-CMS2-pCXR9T. The REG2-BARNASE expression cassette was cloned into theI site of the plasmids Actin1P-CMS2-pCXR9T and OsGEX2P-CMS2-pCXR9T by primer pair REG2P-F/BAR-R to generate TKC1.1 and TKC1.2, respectively.
Insertion of sgRNA transcriptional cassettes into TKC vectors
To generate the plasmids TKC1.1-LAZY1 and TKC1.2-LAZY1, the sgRNA production cassette was inserted into theI site of TKC1.1 and TKC1.2 by overlapping PCR with primer pairs OsU6P-F/OsU6T-R and LAZY1-U6F/LAZY1-U6R. The sgRNA ofwas produced by the rice U6 promoter. The sequence of the rice U6:sgRNA cassette is shown in Supplemental Fig. 1. The details of cloning the sgRNA unit are described below.
TKC plasmid construction with sgRNA transcriptional cassette
The target sequence of genewas selected using the CRISPR-P website tool (Lei et al, 2014). The target sequence of the sgRNA driven by the rice U6 promoter was defined as GNNNNNNNNNNNNNNNNNNNNGG (N refers to any nucleotide) and NGG was the PAM.
Primers for amplifying the sgRNA units were designed as described below. The forward primer was named as X-U6F (X is the gene name. For example, for thegene, the primer was called LAZY1-U6F) and the complete sequence of X-U6F is GNNNNNNNNNNNNNNNNNNNgttttagagctagaaatagcaagtta, where ‘N’ is the forward sequence of the target sequence, and the sequence in lower case is part of the sgRNA core sequence. The reverse primer was named X-U6R, which has a sequence ofMMMMMMMMMMMMMMMMMMMCaacctgagcctcagcgcagc. ‘M’ is the reverse complement sequence of ‘N’ in the target. The sequence in lower case is part of the rice U6 promoter sequence. Boundary primers named OsU6P-F and OsU6T-R were used to amply the entire OsU6pro-sgRNA unit. OsU6P-F and OsU6T-R were partially overlapping with the sequences at theI restriction site in the TKC plasmids.
Two rounds of PCR amplifications were conducted to generate the transcriptional units of sgRNA. The first round of PCR consisted of two PCR reactions (PCR1 and PCR2). Both PCR1 and PCR2 used any plasmids that contain an OsU6pro- sgRNA as template. PCR1 used the primer pair OsU6P-F and X-U6R. PCR2 used the primer pair X-U6F and OsU6T-R. The products of PCR1 and PCR2 were purified and then mixed. The second round PCR (PCR3) used the OsU6P-F/OsU6T-R primer pair with 1 μL mixed products of PCR1 and PCR2 as a template. The purified product from PCR3 was cloned into theI site of TKC vectors by Gibson assembly. All constructs were sequenced using the primers sgRNA-SeqF and sgRNA-SeqR to confirm the sequence identity.
Plant transformation
Both TKC1.1-LAZY1 andTKC1.2-LAZY1plasmids were transformed into calli of Zhonghua 11 through- mediated rice transformation following the protocol (Hiei et al, 1994). T0plants were transplanted to the normal condition rice field and visually scored forphenotypes. The seeds from each individual T0plants were harvested separately.
Characterization of T1 plants
We randomly selected the T1progeny from 11 independent T0plants (#1, #11, #16, #24, #27, #29, #32, #36, #42, #52 and #59) of TKC1.1-LAZY1 and 5 independent T0plants (#3, #4, #5, #17 and #22) of TKC1.2-LAZY1 to determine the efficiency in self-eliminating the transgenes and in editing the target gene. We used two primer pairs to amplify the specific regions of the TKC plasmids. CC-F/CC-R was used to amply part of theandgenes, and BAR-377F/BAR-377R was used to detect the presence of thegene. The genomic DNA from the corresponding T0plants was used as the positive control, while DNA from Zhonghua 11 used as the negative control. ActinM-F/ActinM-R primer pair was used to check the quality of our genomic DNA samples.
LAZY1-GT1 and LAZY1-GT2 were used to amplify part of thegene from 126 TKC1.1-LAZY1and 75 TKC1.2-LAZY1 T1plants. The PCR products were directly sequenced for mutation analysis. For heterozygous or bi-allelic plants, the overlapping peaks were resolved by using the Dsdecode web tool (Xie et al, 2017).
Results
Development of two new TKC vectors
In order to avoid the potential pitfalls of our previous TKC system (He et al, 2018) (named TKC1.0), which used the CaMV35S-CMS2 cassette to kill transgene-containing pollens, we replaced the CaMV35S promoter with two rice promoters. The OsACTIN1p-CMS2 construct was named TKC1.1 and the OsGEX2p-CMS2 was named as TKC1.2 (Fig. 1-A). We kept the REG2-BARNASEcassette, which killed transgene-containingembryos in the T0plants of both TKC1.1 and TKC1.2 (Fig. 1-A).
T0 plants from our new TKC vectors displayed lazy phenotypes
To test the validity of our strategy, we again targeted thegene (Li et al, 2007; Yoshihara and Iino, 2007) (Fig. 1-C), whose loss-of-function mutations result in an increase in the tiller angle (Fig. 1-C). Therefore, we can estimate the efficiency of TKC1.1 and TKC1.2 in generating homozygous or bi-allelicmutants at T0generation. We generated 59 T0plants from the TKC1.1-LAZY1 construct. Among the 59 plants, 31 displayed obviousphenotypes. The efficiency (52.5%) was higher than that of TKC1.0-LAZY1 construct, which had 44.5% (29/65) T0plants with thephenotype. We found that TKC1.2-LAZY1 construct also had a slightly higher efficiency than TKC1.0-LAZY1. About 47.5% (28 out of 59) T0plants from the TKC1.2-LAZY1 had thephenotypes.
New TKC vectors were effective in self-eliminating the transgenes
Seeds from each individual T0plants were harvested (Fig. 1-B). We analyzed T1plants for the presence of the transgenes and mutations in thegene. We showed previously that at least 75% of T1plants harbored the CRISPR/Cas9 construct when the conventional CRISPR/Cas9 constructs were used (He et al, 2018) and that the TKC1.0 construct was effective in eliminating the CRISPR/Cas9 constructs. We analyzed the 126 TKC1.1-LAZY1 T1plants, and all of them did not contain the transgenes. We also found that the 75 TKC1.2-LAZY1 T1plants were also transgene-free. The results indicated that both the TKC1.1 and TKC1.2 vectors are very effective in eliminating transgenic DNA fragments.
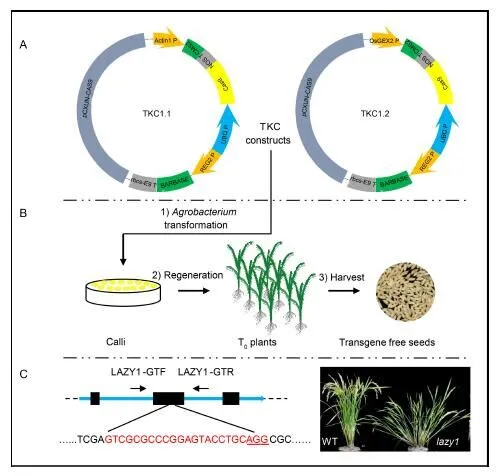
Fig. 1. Transgene killer CRISPR (TKC) vectors enable the isolation of transgene-free CRISPR/Cas9-edited rice plants within a single generation.
A, Schematic presentation of the TKC1.1 and TKC1.2 plasmids. B, Flow chart of the key steps for isolating transgene-free CRISPR/Cas9 edited rice mutants with TKC1.1 or TKC1.2. 1) Delivering the construct to rice calli by-mediated transformation; 2) TKC-containing calli are regenerated to seedlings; 3) Seeds harvested from TKC1.1 or TKC1.2 T0plants are transgene-free. C, Testing the effectiveness of TKC1.1 and TKC1.2 constructs for generating transgene-free editedmutants. CRISPR/Cas9 target sequence is marked in red. The underlined ‘AGG’ is the protospacer adjacent motifsite. The primers LAZY1-GTF and LAZY1-GTR were used for genotyping the T1plants. WT refers to wild-type rice Zhonghua 11, andstands forCRISPR mutant.
Stable transgene-free and edited plants were easily obtained at T1 generation
All of the 126 TKC1.1-LAZY1 T1plants had a mutation near the PAM site of the target sequence (Table 1). Almost all of the 73 TKC1.2-LAZY1 T1plants were either homozygous or bi-allelic at thelocus. However, we found two T1plants from the #17 T0plant that had a WT genotype (Table 2), despite that the #17 T0plant had aphenotype. Our results demonstrated that both TKC1.1 and TKC1.2 vectors were effective for generating targeted editing events.
All of the T1plants generated from TKC1.1-LAZY1 T0plants #1, #27 and #52 were homozygous of the same ‘C’ deletion at thelocus, indicating that the T0plants were already homozygous. Some of the TKC1.1-LAZY1 T0plants such as #11, #29 and #59 produced three genotypes (Table 1), including two types of homozygous and one bi-allelic mutations, revealing the bi-allelic nature of these T0plants. Similarly, some of the T0plants of TKC1.2-LAZY1 including #3 and #4 only produced homozygous T1plants (Table 2). TKC1.2-LAZY1 T1plants from T0plants #3 and #4 showed deletions of ‘C’ and ‘CC’, respectively (Table 2).
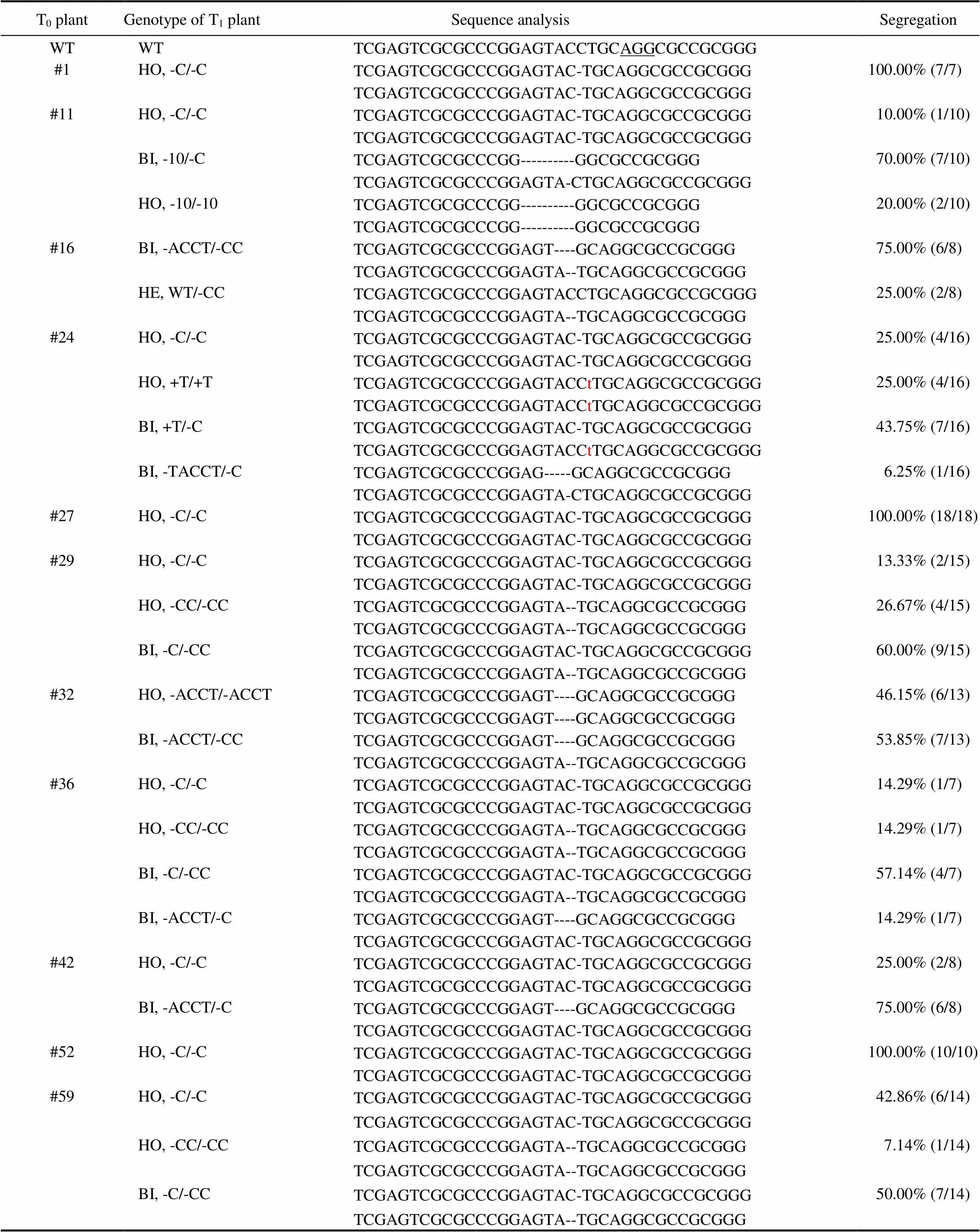
Table 1. Mutations and segregation patterns of TKC1.1-LAZY1 T1 plants.
WT, Wild-type Zhonghua 11 plant; HO, Homozygous mutation; HE, Heterozygous mutation; BI, Bi-allelic mutation.
The underlined ‘AGG’ refers to the protospacer adjacent motif site required for Cas9 cleavage. ‘-’ refers to one base pair deletion. Lowercase letter ‘t’ in red refers to an insertion of a ‘T’. Number of plants for the genotype and the total number of the T1plants from each T0plant analyzed are shown in the parenthesis.
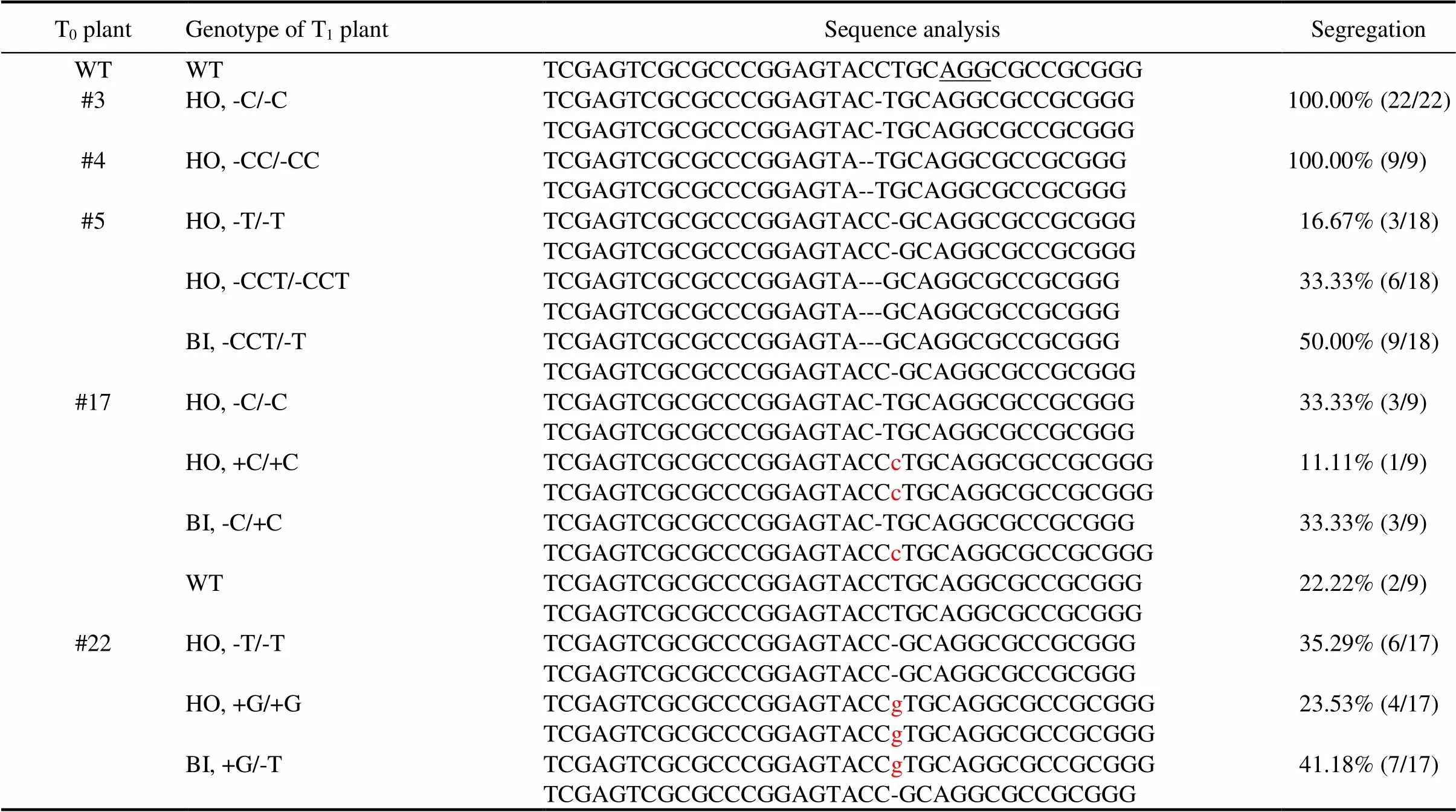
Table 2. Mutations and segregation analysis of the TKC1.2-LAZY1 T1 plants.
Protospacer adjacent motif site AGG is underlined. Zhonghua 11 plants were used as wild-type (WT). ‘+’ refers to base pair insertion. ‘-’ refers to base pair deletion. Lowercase letter ‘c’ and ‘g’ in red refer to insertions of ‘C’ and ‘G’, respectively.
Number of plants for the genotype and the total number of the T1plants from each T0plant analyzed are shown in the parenthesis.
We also noticed some unusual segregation patterns among T1plants, which were observed in our previous TKC1.0 plants as well. There were two types of mutations among the eight T1plants from the #16 TKC1.1-LAZY1 T0plant (Table 1). Six plants were bi-allelic of two mutations: a deletion of ‘ACCT’ and a deletion of ‘CC’. The other two plants deleted a ‘CC’ but were heterozygous (Table 1). Although the #16 TKC1.1- LAZY1 T0plant showedphenotype, it was clear that some types of the T0plant did not completely loseactivity. We also observed a more complex segregation pattern of the T1plants from the TKC1.1-LAZY1 T0plants #24 and #36. Progeny from the T0plant #24 had three types ofmutations: a deletion of ‘C’; an insertion of ‘T’, and a deletion of ‘TACCT’. Whereas the ‘C’ deletion and the ‘T’ insertion appeared bi-allelic in T0plant #24, an unexpected bi-allelic plant with a deletion of ‘TACCT’ and a deletion of ‘C’ was also identified (Table 1). Similar mosaicism was also observed among the T1plants from the TKC1.1-LAZY1T0plant #36.
The segregation patterns of TKC1.2-LAZY1 T1plants (Table 2) were similar to those observed among TKC1.1- LAZY1 T1plants (Table 1). Some of the T0plants such as #3 and #4 only produced homozygousT1plants, and both #5 and #22 should be bi-allelic, whereas another T0plant #17 was apparently mosaic (Table 2).
Discussion
In this study, we reported two new TKC vectors that showed efficient self-elimination of the transgenes and effective gene-editing capacity. The new vectors offered more options for using our TKC technology.
A major advantage of our TKC technology is that it enables unambiguous demonstration of the mosaic nature of the CRISPR/Cas9 edited plants at T0generation in rice (He et al, 2018). T1plants from a single T0plant often displayed more than two types of mutations, which cannot be produced from bi-allelic or heterozygous plants (Tables 1 and 2). T0plants showed mutant phenotypes, however, this cannot guarantee that the locus is bi-allelic or homozygous either (Table 2). For example, the T0plant #17 from TKC1.2-LAZY1 produced wild-type progeny, although it had an obviousphenotype (Table 2). If the CRISPR construct is not removed, it would be very difficult to determine whether the unexpected mutations and segregation patterns are caused by mosaicism or are resulted from continuing Cas9 editing activities. Our results also suggest that genotyping and sequencing T0plants probably is not very meaningful except that such efforts can demonstrate whether the constructs are functional or not.
Our TKC1.0 vector, which expresses thegene under the control of the widely used CaMV35S promoter, is very effective and is probably good enough for most of the gene editing applications. Our new vectors displayed slightly higher editing efficiency based on thephenotypes observed among the T0plants. However, we had shown previously that T0plants without thephenotypes are also often edited, but the mutations such as a deletion of 3 bp did not disrupt the gene functions. Therefore, the slightly increased efficiency of the new vectors may not be very meaningful. We believe that the rice promoters used in the new TKC vectors may provide advantages over TKC1.0 under certain conditions. Because CaMV35S promoter has been widely used in rice and in other plants, many genetic materials may have already contained the CaMV35S promoter, which may interfere with theexpression in TKC1.0. Many plant vectors use the CaMV35Spromoter to drive antibiotic resistance. Moreover, constitutive expression ofunder the control of CaMV35S promoter may negatively affect plant growth, especially for those plants with strong developmental phenotypes when the target gene is edited. The pollen-specific promoter OsGEX2 drivenexpression can further limit any negative impact ofon plant growth and development. The two new vectors presented here provided more options and flexibility for scientists to use our TKC technology.
Acknowledgements
This study was partially supported by Chinese Ministry of Agriculture and Rural Affairs (Grant No. 2018ZX0801003B) and the National Transgenic Science and Technology Program (Grant No. 2016ZX08010002).
SUPPLEMENTAL DATA
The following materials are available in the online versionof this article at http://www.sciencedirect.com/science/ journal/16726308; http://www.ricescience.org.
Supplemental Table 1. Primers used in this study.
Supplemental Fig. 1. Sequence of the rice U6:sgRNAcassette.
Burstein D, Harrington L B, Strutt S C, Probst A J, Anantharaman K, Thomas B C, Doudna J A, Banfield J F. 2017. New CRISPR- Cas systems from uncultivated microbes., 542: 237–241.
Casini A, Olivieri M, Petris G, Montagna C, Reginato G, Maule G, Lorenzin F, Prandi D, Romanel A, Demichelis F, Inga A, Cereseto A. 2018. A highly specific SpCas9 variant is identified byscreening in yeast., 36(3): 265–271.
Cermak T, Baltes N J, Cegan R, Zhang Y, Voytas D F. 2015. High-frequency, precise modification of the tomato genome., 16: 232.
Chu V T, Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, Kuhn R. 2015. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells., 33(5): 543–548.
Cong L, Ran F A, Cox D, Lin S L, Barretto R, Habib N, Hsu P D, Wu X B, Jiang W Y, Marraffini L A, Zhang F. 2013. Multiplex genome engineering using CRISPR/Cas systems., 339: 819–823.
Cook M, Thilmony R. 2012. Thegene promoter confers sperm cell expression in transgenic rice., 30(5): 1138–1148.
Dreissig S, Schiml S, Schindele P, Weiss O, Rutten T, Schubert V, Gladilin E, Mette M F, Puchta H, Houben A. 2017. Live cell CRISPR-imaging in plants reveals dynamic telomere movements., 91(4): 565–573.
Fonfara I, Richter H, Bratovic M, Le Rhun A, Charpentier E. 2016. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA., 532: 517–521.
Gallego-Bartolome J, Gardiner J, Liu W L, Papikian A, Ghoshal B, Kuo H Y, Zhao J M C, Segal D J, Jacobsen S E. 2018. Targeted DNA demethylation of thegenome using the human TET1 catalytic domain., 115(9): 2125–2134.
Gao L, Cox D B T, Yan W X, Manteiga J C, Schneider M W, Yamano T, Nishimasu H, Nureki O, Crosetto N, Zhang F. 2017. Engineered Cpf1 variants with altered PAM specificities., 35(8): 789–792.
Gao X H, Chen J L, Dai X H, Zhang D, Zhao Y D. 2016. An effective strategy for reliably isolating heritable and cas9-freemutants generated by CRISPR/Cas9-mediated genome editing., 171(3): 1794–1800.
Gaudelli N M, Komor A C, Rees H A, Packer M S, Badran A H, Bryson D I, Liu D R. 2017. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage., 551: 464–471.
Gibson D G, Young L, Chuang R Y, Venter J C, Hutchison C A, Smith H O. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases., 6(5): 343–345.
Gilbert L A, Larson M H, Morsut L, Liu Z, Brar G A, Torres S E, Stern-Ginossar N, Brandman O, Whitehead E H, Doudna J A, Lim W A, Weissman J S, Qi L S. 2013. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes., 154(2): 442–451.
Hall T C, Kumpatla S P, Kharb P, Iyer L, Cervera M, Jiang Y, Wang T, Yang G, Teerawanichpan P, Narangajavana J, Dong J. 2001. Gene silencing and its reactivation in transgenic rice.: Rice Genetics IV. World Scientific: 465–481.
He Y B, Wang R C, Dai X H, Zhao Y D. 2017a. On improving CRISPR for editing plant genes: Ribozyme-mediated guide RNA production and fluorescence-based technology for isolating transgene-free mutants generated by CRISPR., 149: 151–166.
He Y B, Zhang T, Yang N, Xu M L, Yan L, Wang L H, Wang R C, Zhao Y D. 2017b. Self-cleaving ribozymes enable the production of guide RNAs from unlimited choices of promoters for CRISPR/Cas9 mediated genome editing., 44(9): 469–472.
He Y B, Zhu M, Wang L H, Wu J H, Wang Q Y, Wang R C, Zhao Y D. 2018. Programmed self-elimination of the CRISPR/Cas9 construct greatly accelerates the isolation of edited and transgene-free rice plants., 11(9): 1210–1213.
Hiei Y, Ohta S, Komari T, Kumashiro T. 1994. Efficient transformation of rice (L.) mediated byand sequence analysis of the boundaries of the T-DNA., 6(2): 271–282.
Hu J H, Miller S M, Geurts M H, Tang W, Chen L, Sun N, Zeina C M, Gao X, Rees H A, Lin Z, Liu D R. 2018. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity., 556: 57–63.
Hua K, Tao X P, Yuan F T, Wang D, Zhu J K. 2018. Precise A∙T to G∙C base editing in the rice genome., 11(4): 627–630.
Huang Y H, Su J Z, Lei Y, Brunetti L, Gundry M C, Zhang X T, Jeong M, Li W, Goodell M A. 2017. DNA epigenome editing using CRISPR-Cas SunTag-directed DNMT3A., 18(1): 176.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna J A, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity., 337: 816–821.
Kleinstiver B P, Prew M S, Tsai S Q, Topkar V V, Nguyen N T, Zheng Z, Gonzales A P, Li Z, Peterson R T, Yeh J R, Aryee M J, Joung J K. 2015. Engineered CRISPR-Cas9 nucleases with altered PAM specificities., 523: 481–485.
Komor A C, Kim Y B, Packer M S, Zuris J A, Liu D R. 2016. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage., 533: 420–424.
Konermann S, Brigham M D, Trevino A E, Joung J, Abudayyeh O O, Barcena C, Hsu P D, Habib N, Gootenberg J S, Nishimasu H, Nureki O, Zhang F. 2015. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex., 517: 583–588.
Lei Y, Lu L, Liu H Y, Li S, Xing F, Chen L L. 2014. CRISPR-P: A web tool for synthetic single-guide RNA design of CRISPR- system in plants., 7(9): 1494–1496.
Li J Y, Sun Y W, Du J L, Zhao Y D, Xia L Q. 2017. Generation of targeted point mutations in rice by a modified CRISPR/Cas9 system., 10(3): 526–529.
Li J Y, Zhang X, Sun Y W, Zhang J H, Du W M, Guo X P, Li S Y, Zhao Y D, Xia L Q. 2018. Efficient allelic replacement in rice by gene editing: A case study of thegene., 60(7): 536–540.
Li P J, Wang Y H, Qian Q, Fu Z M, Wang M, Zeng D L, Li B H, Wang X J, Li J Y. 2007.controls rice shoot gravitropism through regulating polar auxin transport., 17(5): 402–410.
Li S Y, Li J Y, Zhang J H, Du W M, Fu J D, Sutar S, Zhao Y D, Xia L Q. 2018a. Synthesis-dependent repair of Cpf1-induced double-strand DNA breaks enables targeted gene replacement in rice., 69(20): 4715–4721.
Li S Y, Zhang X, Wang W S, Guo X P, Wu Z C, Du W M, Zhao Y D, Xia L Q. 2018b. Expanding the scope of CRISPR/Cpf1- mediated genome editing in rice., 11(7): 995–998.
Li Z X, Zhang D D, Xiong X Y, Yan B Y, Xie W, Sheen J, Li J F. 2017. A potent Cas9-derived gene activator for plant and mammalian cells., 3: 930–936.
Liu X S, Wu H, Ji X, Stelzer Y, Wu X B, Czauderna S, Shu J, Dadon D, Young R A, Jaenisch R. 2016. Editing DNA methylation in the mammalian genome., 167(1): 233–247.
Lu Y M, Zhu J K. 2017. Precise editing of a target base in the rice genome using a modified CRISPR/Cas9 system., 10(3): 523–525.
Mali P, Yang L H, Esvelt K M, Aach J, Guell M, DiCarlo J E, Norville J E, Church G M. 2013. RNA-guided human genome engineering via Cas9., 339: 823–826.
McElroy D, Zhang W G, Cao J, Wu R. 1990. Isolation of an efficientpromoter for use in rice transformation., 2(2): 163–171.
Murovec J, Pirc Z, Yang B. 2017. New variants of CRISPR RNA-guided genome editing enzymes., 15(8): 917–926.
Ren B, Yan F, Kuang Y J, Li N, Zhang D W, Zhou X P, Lin H H, Zhou H B. 2018. Improved base editor for efficiently inducing genetic variations in rice with CRISPR/Cas9-guided hyperactivemutant., 11(4): 623–626.
Stepper P, Kungulovski G, Jurkowska R Z, Chandra T, Krueger F, Reinhardt R, Reik W, Jeltsch A, Jurkowski T P. 2017. Efficient targeted DNA methylation with chimeric dCas9-Dnmt3a-Dnmt3L methyltransferase., 45(4): 1703–1713.
Sun Y W, Zhang X, Wu C Y, He Y B, Ma Y Z, Hou H, Guo X P, Du W M, Zhao Y D, Xia L Q. 2016. Engineering herbicide- resistant rice plants through CRISPR/Cas9-mediated homologousrecombination of acetolactate synthase., 9(4): 628–631.
Tak Y E, Kleinstiver B P, Nunez J K, Hsu J Y, Horng J E, Gong J Y, Weissman J S, Joung J K. 2017. Inducible and multiplex gene regulation using CRISPR-Cpf1-based transcription factors., 14(12): 1163–1166.
Tanenbaum M E, Gilbert L A, Qi L S, Weissman J S, Vale R D. 2014. A protein-tagging system for signal amplification in gene expression and fluorescence imaging., 159(3): 635–646.
Vojta A, Dobrinic P, Tadic V, Bockor L, Korac P, Julg B, Klasic M, Zoldos V. 2016. Repurposing the CRISPR-Cas9 system for targeted DNA methylation., 44(12): 5615–5628.
Wang M G, Lu Y M, Botella J R, Mao Y F, Hua K, Zhu J K. 2017. Gene targeting by homology-directed repair in rice using a geminivirus-based CRISPR/Cas9 system., 10(7): 1007–1010.
Wang Z H, Zou Y J, Li X Y, Zhang Q Y, Chen L T, Wu H, Su D H, Chen Y L, Guo J X, Luo D, Long Y M, Zhong Y M, Liu Y G. 2006. Cytoplasmic male sterility of rice with boro II cytoplasm is caused by a cytotoxic peptide and is restored by two related PPR motif genes via distinct modes of mRNA silencing., 18(3): 676–687.
Weinhold A, Kallenbach M, Baldwin I T. 2013. Progressive 35S promoter methylation increases rapidly during vegetative development in transgenicplants., 13: 99.
Wilkinson J E, Twell D, Lindsey K. 1997. Activities of CaMV 35S and nos promoters in pollen: Implications for field release of transgenic plants., 48(2): 265–275.
Xie X R, Ma X L, Zhu Q L, Zeng D C, Li G S, Liu Y G. 2017. CRISPR-GE: A convenient software toolkit for CRISPR-based genome editing., 10(9): 1246–1249.
Yan F, Kuang Y J, Ren B, Wang J W, Zhang D W, Lin H H, Yang B, Zhou X P, Zhou H B. 2018. Highly efficient A∙T to G∙C base editing by Cas9n-guided tRNA adenosine deaminase in rice., 11(4): 631–634.
Yoshihara T, Iino M. 2007. Identification of the gravitropism- related rice geneand elucidation of-dependent and -independent gravity signaling pathways., 48(5): 678–688.
Yu C C, Wang L L, Xu S L, Zeng Y F, He C L, Chen C, Huang W C, Zhu Y G, Hu J. 2015. Mitochondrial ORFH79 is essential for drought and salt tolerance in rice., 56(11): 2248–2258.
Zetsche B, Gootenberg J S, Abudayyeh O O, Slaymaker I M, Makarova K S, Essletzbichler P, Volz S E, Joung J, van der Oost J, Regev A, Koonin E V, Zhang F. 2015. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system., 163(3): 759–771.
Zetsche B, Heidenreich M, Mohanraju P, Fedorova I, Kneppers J, DeGennaro E M, Winblad N, Choudhury S R, Abudayyeh O O, Gootenberg J S, Wu W Y, Scott D A, Severinov K, van der Oost J, Zhang F. 2016. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array., 35: 31–34.
Zhong Z H, Zhang Y X, You Q, Tang X, Ren Q R, Liu S S, Yang L J, Wang Y, Liu X P, Liu B L, Zhang T, Zheng X L, Le Y, Zhang Y, Qi Y P. 2018. Plant genome editing using FnCpf1 and LbCpf1 nucleases at redefined and altered PAM sites., 11(7): 999–1002.
Zhou Y X, Wang P, Tian F, Gao G, Huang L, Wei W S, Xie X S. 2017. Painting a specific chromosome with CRISPR/Cas9 for live-cell imaging., 27: 298–301.
(Managing Editor: Wang Caihong)
Copyright © 2019, China National Rice Research Institute. Hosting by Elsevier B V
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/)
Peer review under responsibility of China National Rice Research Institute
http://dx.doi.org/10.1016/j.rsci.2018.11.001
25 September 2018;
26 November 2018
Zhao Yunde (yundezhao@ucsd.edu)
杂志排行

Rice Science的其它文章
- Production of Two Elite Glutinous Rice Varieties by Editing Wx Gene
- Development and Application of CRISPR/Cas System in Rice
- Rapid Creation of New Photoperiod-/Thermo-Sensitive Genic Male-Sterile Rice Materials by CRISPR/Cas9 System
- CRISPR/Cas9-Mediated Adenine Base Editing in Rice Genome
- Targeted Mutagenesis of NAC Transcription Factor Gene, OsNAC041, Leading to Salt Sensitivity in Rice
- Characterization and Evaluation of OsLCT1 and OsNramp5 Mutants Generated Through CRISPR/Cas9-Mediated Mutagenesis for Breeding Low Cd Rice