基于MapReduce 的多样本基因鉴定并行优化模型
2019-03-05刘佳俊胡大裟蒋玉明
刘佳俊,胡大裟,蒋玉明
(四川大学计算机学院,成都610065)
0 引言
基因测序与变异鉴定技术在基因研究领域中发挥着非常重要的作用。随着下一代测序技术[1]诞生,测序成本下降,运用重测序研究群体遗传变异成为一种可行方法。然而随着测序深度和样本数目剧增,面对海量的基因序列,鉴定并寻找单核苷酸多态性(Single Nucleotide Polymorphism,SNP)[2-3]位点需要大量的计算资源和时间,基因数据的分析速度成为研究的瓶颈之一。在保证鉴定的高精度前提下,如何提高基因鉴定的速度,降低时间成本,已成为基因鉴定中的关键技术之一。
近年来,已经有许多学者将基因测序技术与计算机软件研究相结合,如:BWA[4]软件在短序列的分析应用中能达到较高的准确率。Heng Li[5]使用无衍生物最小化(Minimization Without Derivatives,MWD)等方法[6]估算等位基因频率,并提出了一种突变识别(variant calling)的统计学框架。Aaron McKenna 等[7]提出了一个使用分布式和共享内存并行化的结构化编程框架—基因组分析工具包(The Genome Analysis Toolkit,GATK)。Dries Decap 等[8]则根据MapReduce[9]编程模型在多节点环境下实现并行分布式内存计算。M.C.Schatz[10]使用随机微卫星扩增多态DNA(RMAP)建模,提出一种通过Hadoop 实现的高度敏感并行种子扩增读取映射算法。
面对日益增长的基因数据,多线程的基因分析工具出现了计算耗时过长的问题。而在实际应用中,往往又需要进行多个样本的SNP 鉴定,以SNP 鉴定中主要使用的工具SAMtools 为例,对一个约为4GB 的测序样本,鉴定单个样本的计算耗时约为三至四天;而当样本数量增加至10 个时,计算时间随之增至约一个月;当样本数量增加至200 个,计算时间至少需要两个月。
本文设计了一个基于SPMD(Single Program,Multiple Data)[11]模式的并行化模型,在高性能计算平台上结合传统鉴定工具的优点,根据MapReduce 框架将数据划分和映射,把计算任务拆分并利用多节点进行计算,并实现多样本的并行计算,充分利用计算平台多节点多CPU 资源,解决传统工具在高性能计算平台上性能较低的问题,提高遗传变异鉴定的计算速度以及加速变异检测过程。
1 多节点并行化模型
MapReduce 是用于处理大量数据的一种支持并行化的分布式模型,MapReduce 主要有Map(映射)和Reduce(归约)两部分组成。Map 函数根据输入基因数据,划分出若干个参考序列——样本序列组合,Reduce函数收集包含相同序列区间并对相应位点进行单核苷酸多态性鉴定。
SNP 鉴定主要是通过将参考序列和对应的若干个样本序列进行组合,对每个基因位点的匹配程度进行统计。假设F 为某一染色体参考序列,Si为样本代号i在该染色体上的对应序列,则{Si,Sj,…,Sn}是n 个样本在该染色体上的样本序列集合。将F 和{Si,Sj,…,Sn}的参考序列——样本序列进行组合;假设参考序列上的某一基因位点为GF,GS是样本序列上与GF对应位置的位点,将包含GF位点信息的read 序列与GS进行匹配等计算统计,评估GS处发生SNP 的可能性。
1.1 多样本多节点的并行计算模型
Map 函数将参考序列和样本序列分别进行划分,得到若干个Fk和对应的{Si-k,Sj-k,…,Sn-k}的参考序列——样本序列组合,然后将具有相同序列区间的多个样本数据进行合并,再由Reduce 函数收集和归约并计算这些参考序列——样本序列组合的基于Phred 的质量得分Q(Phred-like Quality Scores),经序列对齐等相关处理,最后过滤筛选,输出SNP 候选位点信息[12]。各个节点上的计算流程如图1 所示。
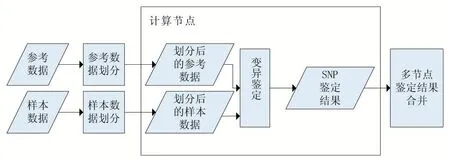
图1 各计算节点上的计算流程
(1)参考数据:参考的基因组数据,包含模板生物序列的DNA 或者蛋白质序列信息。
(2)样本数据:实际样本测序得到的基因数据,包含了多个read 比对信息,用于基因鉴定与分析。
(3)参考数据划分:以计算节点数量和参考数据大小为参考,将参考序列分割为合理大小用于并行计算。
(4)样本数据划分:以参考分割程序的划分地址为参考,将样本序列分割为合理大小用于并行计算。
(5)变异鉴定:将划分后的参考数据和样本数据映射匹配,对序列中的每一个位点进行SNP 概率估计。
(6)多节点鉴定结果合并:将多个计算节点生成的结果文件按照划分地址的顺序进行排列与组合。
1.2 任务分配方案
动态负载优先(Dynamic Load Priority,DLP)方案首次任务分配时随机选择若干节点,通过控制主机进行主动探测,获得当前子节点的负载情况和计算进度,再对子节点的负载和资源量进行优先级判定。在计算平台上有若干计算节点N={N1,N2,…,Nn},其中节点Ni的空余资源为Rf,假设节点Ni上已有k 个任务正在计算,其中任务Tj占用的资源为Rj,对应的计算进度百分比为Sj;当前进行任务分配的计算资源请求为R,则节点Ni的分配优先级Pi为:

当控制主机进行再分配时会结合当前各节点资源的优先级来指派任务,选择优先级别高的节点进行再分配。
1.3 Map函数和Reduce函数
根据SNP 鉴定中数据文件的种类[13],进行数据划分的Map 处理可以分为参考序列和样本序列两种:参考序列的Map 处理首先需要按照染色体种类进行拆分,然后通过设置并行数量,对拆分后的单染色体参考序列,进行平均分割操作以实现各节点负载均衡;样本序列的Map 处理会根据参考序列Map 的分割结果,提取并划分对应区间的样本序列。
Map 函数的输入为(key,value)键值对,其中key 为参考序列每个分割区间的起始地址,value 为对应区间的参考序列。Map 函数将参考基因组根据区间长度或者测序深度进行平均划分,保证每段数据量相近。Map函数完成数据划分后,生成对应的(key,value)文件存储在磁盘上,进入下一步计算操作。
参考序列的Map 函数描述如下:
函数1 参考序列的Map 函数。
输入:参考序列数据。
输出:参考序列键值对。
Step1 控制节点将输入的参考序列文件按照其中数据的染色体名称进行分离,得到多个单染色体参考序列文件。
Step2 控制节点获取指定的划分数量,确定进行划分的参考序列的最小阈值。对上述每个参考序列进行Step3 到Step4 的划分操作。
Step3 获取数据文件的大小,选择根据划分数量进行平均划分或者根据测序深度进行划分。
Step4 建立起始地址——对应划分区间数据的键值对,输出至硬盘上并结束。
参考序列的Map 函数运行在控制节点上,时间开销取决于输入的参考序列大小,参考序列主要由字符组成,遍历的速度较快。控制节点统计输出的起始地址——划分数据键值对数量,根据DLP 方案进行任务分配。在每个计算节点均需上执行样本序列——Map函数以及相关处理。
在参考序列的Map 函数完成参考序列的分割后,每个计算任务并行进行每个任务的排序和提取等操作。在提取对应区间内的序列信息时,可能会导致在区间两端的信息出现缺失,从而影响鉴定结果。为了减小和避免这种情况,在提取序列信息时额外增加提取区间两端的范围,可确保序列信息的完整。
样本序列的Map 函数描述如下:
函数2 样本序列的Map 函数。
输入:样本序列数据以及起始地址——划分数据键值对。
输出:样本序列键值对。
Step1 计算节点把样本序列数据根据参考序列中的染色体进行分离。
Step2 对每个染色体的样本序列进行区间统计,判断当前地址区间内是否有相关的读长read 信息[14],如果区间内包含相关信息则根据参考序列中的地址区间进行划分,划分时额外增加的区间长度为l,通过根据各个read 的长度和出现的频次的加权计算,如公式(2);如果没有则输出空白文件。

式(2)中,Li为编号为i 的read 序列的长度,Ni为编号为i 的read 序列出现的次数。
Step3 输出样本序列键值对至硬盘上,完成参考序列-样本序列组的划分映射并结束。
在经过Map 函数划分后,计算节点对输出的参考序列——样本序列组合进行区间地址的校验,校验通过后使用Reduce 函数归约并进行SNP 鉴定计算。若校验未通过重新进行Step2。
Reduce 函数描述如下:
函数3 Reduce 函数。
输入:参考序列——样本序列组合。
输出:SNP 鉴定结果文件。
Step1 计算节点多线程读入多个起始区间的参考序列,并收集所有样本中具有相同区间的样本序列。其中每个线程读入一个计算单元数据,即一个参考序列和与其对应的若干个样本序列。
Step2 对每一组鉴定数据单元,计算序列中的碱基位点的等位基因的先验概率,根据概率选出最有可能变异的位点。
Step3 计算节点将SNP 结果输出至硬盘并结束。
控制节点会持续询问所有计算节点直到完成SNP鉴定,然后控制节点把所有结果根据划分的地址顺序进行合并。
2 实验测试与结果分析
实验测试有2 台曙光A840R-G,4×AMD 6172 12核CPU,512G 内存,共4 个计算节点,操作系统为Linux version 2.6.32-431.el6.x86_64;Red Hat Enterprise Linux Server release 6.5,作业管理系统为LSF。
实验数据以某地某品种(系)烟草材料为作为测试样本,参考序列为随机选择的其第17 号染色体数据,样本序列则为该品种(系)烟草的128 个样本测序数据,每个样本的文件大小约为4GB。实验主要分析样本数和并行数对计算时间的影响,以SAMtools 工具在单节点的计算时间为对比。第一部分实验,固定计算的样本规模,同时改变参与计算的节点数量;第二部分实验,固定每个节点所计算的样本数量,通过增加节点数量,分析计算效率的变化。
采用本文提出的并行优化模型在多节点上设置并行数依次为2 组、3 组、5 组、10 组、15 组、20 组、30 组、50 组、100 组,其中,每组并行数再分别计算样本数1个、2 个、4 个、8 个、16 个、32 个、64 个、128 个,每个样本的大小约4GB。通过上述计算,以运算时间和加速比[15]两个指标来评价本文模型,统计运算时间如图2 所示,加速比如图3 所示。

图2 样本数和并行数对计算效率的影响
由图2 可看出:在并行数保持不变的情况下,样本数与计算时间成正比,即随着样本数的增多,排序等操作所需遍历的文件总大小也在增大,平均每个基因位点的堆叠深度在增加,呈现出计算时间成倍增加;而在样本数保持不变的情况下,在并行数小于20 时,增加并行数可显著降低计算时间,即通过实现并行化,增加并行计算数量,计算时间会呈现明显下降的趋势。
当样本数小于16 个时,多组数据相互对比,计算时间差距并不明显,使用并行化加速效果并不明显,在实际应用中可以考虑使用传统计算方法计算,避免不必要的计算误差;但当样本数为16 个至128 个时,采用并行加速模型可明显增加提高计算效率。
由图3 可看出随着并行数的增加,加速比在并行数20 以内呈现明显上升趋势,说明增加并行数可有效地减小每个任务的计算时间,从而减少总体计算时间;加速比在并行数量30 到50 这个区间出现缓慢上升甚至持平的趋势,这是因为当每个节点的计算时间缩小到一定程度后,SNP 鉴定的时间主要由相对固定耗时的I/O 操作效率所确定,并且已经相当接近,所以导致加速比不再明显增加;当并行数在50 以上后,加速比将保持恒定。由图2 和图3 综合也说明并行数量并不是并非简单的越大越好,而是与样本数有直接关系。
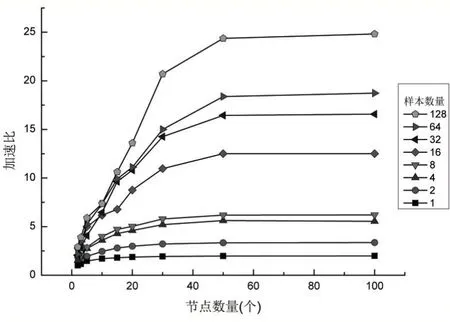
图3 样本数和并行数对多节点计算加速比的影响
3 结语
针对基因变异鉴定耗时较大的问题,通过MapReduce 框架模型将计算任务划分,在多节点上实现并行加速计算,提出了一种针对多样本的多节点并行化优化算法,在海量基因数据的计算与分析中具有广泛适用性。
通过SNP 鉴定的计算实验发现,样本数平均每增加一倍,计算时间也平均增加约一倍;通过增加并行数加速计算,可使计算时间明显下降:并行数平均每增加一倍,计算时间平均减少约32%,加速比平均上升约46%。验证了该优化算法是有效的。同时,并行计算的加速效果受样本数和并行数影响,在小数据量或多余并行下无法显示其优越性,实际计算中应根据样本数和硬件条件,合理规划才能获得更有效的加速效果。
