高效液相色谱法测定枇杷叶膏中绿原酸含量
2019-03-01辜俊鑫张学霞魏晓玲王成琼谯春燕
辜俊鑫 ,辜 瑜 ,张学霞 ,魏晓玲 ,王成琼 ,曾 容 ,谯春燕
(1.北京同仁堂科技发展成都有限公司,四川 成都 610100; 2.中国科学院上海药物研究所,上海 201203)
枇杷叶膏收载于2015年版《中国药典(一部)》,由 枇杷叶经水煮浓缩加炼蜜或蔗糖溶化,再浓缩到规定的相对密度即得,具有清肺润燥、止咳化痰的功效。其质量标准中的检验项目有性状、相对密度、不溶物、装量及微生物限度,无成分的定量项,不能客观地反映产品的质量控制情况。为解决枇杷叶膏生产过程中的枇杷叶前处理和提取,并开发出用于提取车间申报的定量分析方法。前期分析了熊果酸、齐墩果酸[1]、儿茶素、苦杏仁苷、槲皮素、山柰素[2-3]和绿原酸[4],发现有的成分含量太低,不适合定量分析,有的成分在不同药材中的重复性不好,时有时无,最终确定以绿原酸作为定量指标。本研究中采用高效液相色谱法测定枇杷叶膏中的绿原酸含量,作为其质量控制指标之一。现报道如下。
1 仪器与试药
1.1 仪器
Agilent1260型高效液相色谱仪(安捷伦科技<中国>有限公司);T9S紫外-可见分光光度计(北京普析通用仪器有限责任公司);MS205DU型电子天平(梅特勒-托利多公司);AS10200B型超声波处理器(天津奥特赛恩斯仪器有限公司)。
1.2 试药
甲醇(分析纯,成都市科龙化工试剂厂);乙腈(色谱纯,Fisher公司);磷酸(分析纯,天津市科密欧化学试剂有限公司);水为自制二次重蒸馏水;绿原酸对照品(中国食品药品检定研究院,批号为110753-201716,含量为99.3%);枇杷叶膏(市售品,批号分别为17132754,17132755,17132756)。
2 方法与结果
2.1 色谱条件与系统适用性试验
色谱柱:Amethyst C18-H 柱(250 mm ×4.6 mm,5 μm);流动相:乙腈 - 0.4% 磷酸溶液(10 ∶90);检测波长:327 nm;柱温:30 ℃ ;流速:1.0 mL /min;进样量:10 μL。在拟订色谱条件下绿原酸的分离度在 3.0以上,对称因子为1.02,理论板数以绿原酸峰计不低于15 000。以峰面积外标法定量。色谱图见图1。
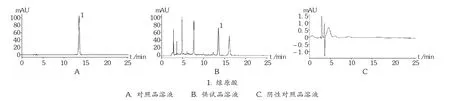
图1 高效液相色谱图
2.2 溶液制备
称取枇杷叶膏4.2 g,置100 mL容量瓶中,加入甲醇 -水(50 ∶50)约 80 mL,超声溶解 30 min,放冷至室温,再用甲醇 -水(50∶50)定容至刻度,摇匀,滤过,取续滤液,即得供试品溶液。取绿原酸对照品适量,精密称定,加甲醇 -水(50 ∶50)制成每 1 mL 含 40 μg的对照品溶液。按制备工艺方法制备不含枇杷叶的阴性样品,按上试品溶液制备方法制备阴性对照品溶液。
2.3 方法学考察
阴性干扰试验:分别取对照品溶液、供试品溶液和阴性对照品溶液各适量,按拟订色谱条件各进样10 μL测定。记录色谱图。可见,供试品溶液色谱图中,在与对照品溶液相应位置处有相应色谱峰,阴性对照无干扰。
线性关系考察:取 10.32 mg经五氧化二磷(P2O5)干燥12 h后的绿原酸对照品,精密称定,置100 mL容量瓶中,用甲醇-水(50∶50)超声溶解并定容至刻度,作为绿原酸对照品贮备液。分别吸取绿原酸对照品贮备液2.0,3.0,4.0,5.0,6.0,7.0 mL,置于 10 mL 量瓶中,加甲醇 -水(50 ∶50)制成质量浓度分别为 20.495 52,30.743 28,40.991 04,51.238 8,61.486 56,71.734 32 μg /mL 的溶液,按拟订色谱条件测定峰面积。以峰面积(Y)为纵坐标、绿原酸进样量(X)为横坐标进行线性回归,得回归方程 Y=3 149.4 X+8.164 1,r=0.999 9(n=6)。结果表明,绿原酸进样量在 0.204 955 2 ~ 0.717 343 2 μg 范围内与峰面积线性关系良好。
精密度试验:取同一对照品溶液,精密吸取10 μL,重复进样 6次,测定峰面积。结果的 RSD为 0.97%(n=6),表明仪器精密度良好。
重复性试验:取同一批样品6份,依法制备供试品溶液并测定绿原酸含量。结果的 RSD为1.2%(n=6),表明方法重复性良好。
稳定性试验:取同一供试品溶液,于室温条件下放置10 h,每隔2 h进样1次,测定峰面积。结果的 RSD为0.83%(n=6),表明供试品溶液在10 h内稳定。
加样回收试验:取绿原酸对照品10.65 mg,精密称定,置100 mL容量瓶中,用甲醇-水(50∶50)超声溶解并定容至刻度,摇匀,作为对照品溶液(绿原酸质量浓度为 105.754 5 μg /mL);精密称取已知含量的样品(批号为17132754)适量,共6份,分别加入上述对照品溶液6.0,6.0,12.0,12.0,18.0,18.0 mL,依法制备供试品溶液,按拟订色谱条件进行测定,计算回收率。结果见表1。
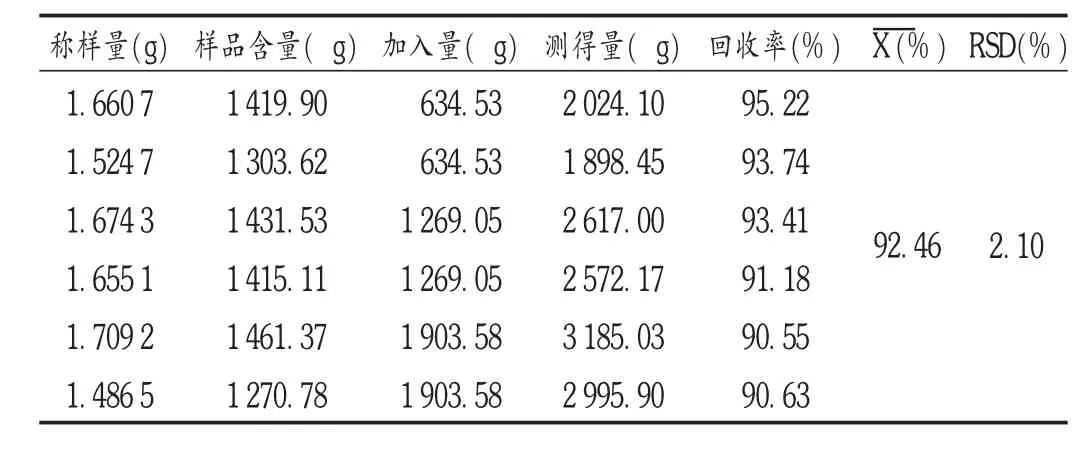
表1 绿原酸加样回收试验结果(n=6)
2.4 样品含量测定
取3批样品,依法制备供试品溶液,并精密称取绿原酸对照品适量,同法制成质量浓度为40 μg/mL的对照品溶液,精密吸取对照品溶液与供试品溶液各10 μL,注入液相色谱仪,依法测定,用外标法计算含量。结果见表2。

表2 3批样品中绿原酸含量测定结果(mg/g,n=3)
3 讨论
3.1 色谱条件选择
在选择检测波长时,取绿原酸对照品溶液,用甲醇-水(50∶50)为空白溶剂,经紫外-可见分光光度计扫描,绿原酸在328 nm波长下有最大吸收,参考《中国药典》金银花中绿原酸的检测波长为327 nm[5],故确定327 nm作为绿原酸的检测波长。曾以乙腈-0.4%磷酸溶液(13∶87)为流动相,但样品分离度和主峰形不好;用乙腈-0.4%磷酸溶液(10∶90)为流动相时,主峰形对称,分离效果好。由于枇杷叶膏很黏稠,用甲醇-水(50∶50)超声溶解时,不定时振摇有助于样品溶解;比较超声20 min和30 min的效果发现,超声30 min时提取完全,故最终采用甲醇-水(50∶50)超声溶解30 min。方法学考察结果表明,所建立的方法回收率、精密度、重复性均符合2015年版《中国药典(四部)》药品质量标准分析方法验证指导原则的相关规定[6],且操作简便、快捷,本方法的建立,可弥补现行药典标准中无成分定量的现状,有利于更好地提高和控制枇杷叶膏的药品质量。
3.2 不足
枇杷叶中绿原酸受各地环境不同、生长时间长短、采收加工时间不同、加工工艺不同影响,其含量会有差异,导致成品枇杷叶膏中绿原酸的含量差异较大。故未对枇杷叶膏中绿原酸的含量做出建议值,如要规定其含量值,应更多地考察和研究枇杷叶在不同产地、不同加工时间段、不同生产工艺对绿原酸的影响,同时比较不同厂家生产的枇杷叶膏的绿原酸含量及绿原酸在枇杷叶膏中的稳定性,为确定一个科学、有效、客观的含量值,提供更多、更全面、更公正的数据。
本研究中还发现,与绿原酸面积归一百分比相近的还有3个主要成分峰,如能确定其中1个成分峰,使用2个成分指标定量,将更有利于提高枇杷叶膏质量标准。
