阻塞型睡眠呼吸暂停合并高血压病人血清3-NT、NO水平变化及其相关性分析
2019-02-28
阻塞型睡眠呼吸暂停(obstructive sleep apnea,OSA)是最常见的一种睡眠障碍性疾病,主要表现为睡眠打鼾并伴有呼吸暂停和/或呼吸表浅、夜间反复低氧血症、高碳酸血症和睡眠呼吸紊乱。流行病学和临床研究结果都证实OSA可累及多系统并造成多器官损害,是高血压、冠心病、充血性心力衰竭、心律失常、糖尿病、脑卒中等多种疾病的独立危险因素。流行病学调查显示,OSA的总体患病率在:成年男性为3%~7%,成年女性为2%~5%[1]。50%~92%的OSA病人合并有高血压,而30%~50%的高血压病人同时伴有OSA[2-3]。OSA导致高血压的机制可以从交感神经机制、肾素-血管紧张素-醛固酮(RAAS)系统的激活、氧化应激、系统炎症反应、胰岛素抵抗等方面来解释。3-硝基酪氨酸(3-NT)、一氧化氮(NO)均是氧化应激的产物,而慢性间歇低氧是OSA的重要病理生理特征。Moya等[4]的研究显示,由NO与超氧阴离子自由基(O2-)反应导致的过氧亚硝酸盐形成是慢性间歇低氧诱发的颈动脉体(CB)化学感受器反应增强和高血压形成的一个关键步骤。过氧化亚硝酸离子(ONOO-)水平增加导致蛋白质的酪氨酸残基变成3-NT[5]。有研究报道,低心血管疾病危险因素的OSA病人NO有效性的减少与微循环内皮细胞过氧亚硝酸盐生成的增加有关[6]。NO是由一氧化氮合酶(NOS)催化L-精氨酸而产生,是内源性NO的唯一来源,因此,NO与NOS活性相关。Varadharaj 等[7]研究发现,内皮型一氧化氮合酶(eNOS)解偶联是OSA病人NO利用度降低和内皮O2-来源生产过剩的原因,eNOS是OSA诱导血管内皮功能障碍的新途径。OSA促进高血压发生发展的确切机制尚未阐明,本研究测定OSA合并高血压病人血清3-NT、NO水平,探讨慢性间歇低氧条件下高血压病人3-NT、NO的变化及发病的可能机制,为OSA合并高血压的病理生理机制提供更充分的理论依据,进一步阐明OSA与高血压之间的关系。
1 资料与方法
1.1 一般资料 选取2016年7月—2017年7月于我科睡眠室行多导睡眠检测而且未进行持续气道正压(CPAP)呼吸机治疗者87例,其中男47例,女40例;年龄18~70岁;其中健康体检正常者(对照组)16名,OSA(OSA组)29例,高血压(高血压组)17例,OSA合并高血压组(OSA合并高血压组)25例。所有研究对象均接受夜间至少7 h的多导睡眠呼吸监测(PSG),监测当天禁饮茶、咖啡、酒及服用镇静药物等,近1个月未服用过叶酸、维生素B6(VitB6)、维生素B12(VitB12)、雌激素等药物,监测前1周戒烟酒,口服降压药者停药1周,且高血压发生在OSA之后。排除标准:肾源性、内分泌等继发性高血压;急慢性感染性疾病;冠心病、心肌梗死、心力衰竭;严重肾脏疾病;慢性呼吸系统疾病;心脑血管疾病;糖尿病、甲状腺功能亢进等内分泌疾病;肿瘤及严重脏器功能衰竭;结缔组织疾病;近期有手术、创伤及其他严重应激病史;精神疾病。
1.2 研究方法
1.2.1 多导睡眠监测 入选者均行夜间不低于7 h 多导睡眠监测,测量身高、体重、颈围、腹围、臀围、睡前及晨起血压。计算体质指数(BMI),记录呼吸暂停指数(apnea-hypopnea index,AHI)、夜间最低血氧饱和度、最长呼吸暂停时间,用Epworth嗜睡量表(ESS)进行嗜睡评分。根据2014年APC发表的《成人阻塞型睡眠呼吸暂停诊断临床实践指南》[8]诊断OSA:AHI≥15次/h,伴或者不伴临床症状;或AHI≥5次/h,伴有临床症状。
1.2.2 血压测量 血压测量采用校正的汞柱式血压计,在标准状态下测量,即同一时间、同侧臂、同一血压计、由同一人测量。重复测血压3次,间隔2 min,取平均值,测量病人睡前及晨起血压。高血压诊断根据2010年《中国高血压防治指南》[9]中的诊断标准:诊室血压≥140/90 mmHg(1 mmHg=0.133 kPa),家庭血压≥135/85 mmHg,24 h动态血压均值≥130/80 mmHg。
1.2.3 血清3-NT、NO测定 所有研究对象在多导睡眠监测结束次日晨起空腹静卧5 min后抽取肘静脉血5 mL,不抗凝,3 000 r/min离心10 min,留取上层血清,-80 ℃冰箱保存。采用酶联免疫吸附法(ELISA)检测血清3-NT,采用化学发光法测定血清NO,试剂盒均购自武汉贝茵莱生物科技有限公司,严格按照试剂盒要求操作。

2 结 果
2.1 各组3-NT、NO水平比较 与对照组相比,OSA组、高血压组、OSA合并高血压组血清3-NT明显升高,血清NO水平明显降低,差异均有统计学意义(P<0.05)。与OSA组、高血压组相比,OSA合并高血压组血清3-NT升高,血清NO水平降低,差异均有统计学意义(P<0.05);OSA组与高血压组血清3-NT、NO水平比较差异无统计学意义(P>0.05)。详见表1。

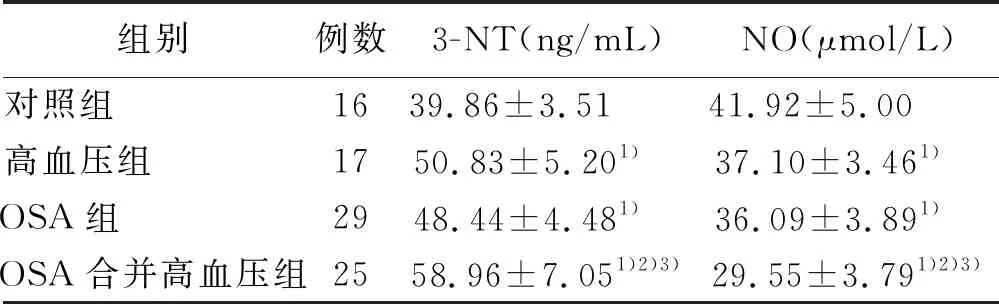
组别例数3-NT(ng/mL)NO(μmol/L)对照组 1639.86±3.51 41.92±5.00 高血压组 1750.83±5.201) 37.10±3.461) OSA组 2948.44±4.481) 36.09±3.891) OSA合并高血压组2558.96±7.051)2)3)29.55±3.791)2)3)
与对照组比较,1)P<0.05;与高血压组比较,2)P<
0.05;与OSA组比较,3)P<0.05
2.2 Pearson直线相关分析 Pearson直线相关分析显示,3-NT与NO呈负相关(r=-0.524,P<0.001)。
3 讨 论
OSA是高血压的独立危险因素[10],氧化应激是OSA导致高血压的重要机制之一。氧化应激是指活性氧簇(ROS)/活性氮(RNS)的产生超过了机体抗氧化的抵抗能力,一般是ROS/RNS产生过多和/或抗氧化能力受损导致机体内氧化及抗氧化能力的失衡[11]。研究证实,OSA病人体内存在严重的氧化应激,OSA合并高血压病人有更明显的氧化应激[12-13]。Schulz等[14]研究发现,慢性间歇性低氧(CIH)条件下大鼠的动脉血压明显增高,NADPH氧化酶2(NOX2)敲除阻断了CIH诱发的高血压的发展。
氧化应激过程中产生的大量ROS与NO和/或NO衍生的RNS发生生物化学反应,生成OONO-,使细胞内蛋白质中的酪氨基残基或者游离的酪氨酸硝化成3-NT[15]。3-NT作为反映机体氧化应激和硝化应激新的生物指标,有研究发现CIH可以增加CB中3 -NT的免疫反应,这与CB化学感受器对缺氧的反应增加相关,且暴露于CIH 7~21 d的大鼠体内3-NT免疫反应积累量随着时间而增加[16]。本研究显示单纯OSA组、OSA合并高血压组的3-NT水平明显高于对照组,证实了慢性间歇低氧条件下硝基化应激水平明显增高。而且Jelic等[17]研究发现,OSA组经过CPAP治疗后氧化应激标志物3-NT较治疗前明显下降,氧化应激水平下降,促进了血管内皮的修复。高血压病人体内抗氧化物质被大量消耗致使抗氧化能力降低,不能有效地清除过多的超氧化物,氧化与抗氧化能力失衡,导致了血管的氧化或硝基化应激损伤,引起血管损伤,最终导致高血压的发生。本研究结果显示,高血压组、OSA合并高血压组NO水平明显低于对照组,提示高血压组、OSA合并高血压组氧化应激损伤加重。有研究证实,高压病病人血清3-NT水平明显升高并与病情进展程度密切相关[18]。其可能的机制为:3-NT通过激活人动脉平滑肌细胞(aorticsmooth- muscle cell,AoSMC)的细胞外信号调节酶1和2(ERK 1/2)信号通路,以及上调与细胞迁移紧密相关因子的mRNA及蛋白表达,同时增加ROS的生成,从而促进AoSMC的迁移[19],使血管平滑肌松弛,动脉血管扩张,从而导致高血压的发生。Moya等[4]研究发现,3-NT能通过蛋白残留物的硝基化促进CB化学感受器反应的增强,这反过来促进高血压的发生,而ONOO-清除剂能有效阻止CB化学感受器反应的增加和逆转慢性间歇低氧诱发的高血压。本研究结果显示,OSA合并高血压组硝基化应激水平较OSA组、高血压组有更高水平的表达,差异有统计学意义,可见慢性间歇低氧诱发的高血压依赖于ONOO-的形成,为今后慢性间歇低氧诱发高血压的治疗提供新的治疗方向。但硝基化应激与慢性间歇低氧条件下高血压之间的具体机制目前还不是十分清楚,准确的机制还需进一步研究。
NO作为传递生命信息的第一信使,是独立于交感神经系统和肾素-血管紧张素系统之外的血压调节因子,具有强烈的血管扩张作用,是最重要的血管舒张因子。本研究结果显示,高血压组较对照组有更低水平的NO表达,可能机制为:NO在内皮细胞NOS的催化下生成,合成后能迅速扩散到邻近的血管平滑肌细胞,与细胞内的特殊受体结合后激活鸟苷酸环化酶(GC)产生环磷酸鸟苷(cGMP),选择性破坏血管平滑肌上cGMP激活的蛋白激酶、水解磷酸二酯酶和调节离子通道,可抑制Ca2+及与Ca2+相关的离子泵,从而干扰血管的舒缩功能,使血管平滑肌松弛、血管扩张。OSA病人反复的间歇低氧和阻塞窒息,窒息时胸腔内负压增大导致静脉回流增加,从而刺激血管内皮释放过多的缩血管活性物质,而舒血管活性物质NO则分泌减少,导致缩血管因子作用占优势,血管舒缩比例失衡,从而造成血管内皮细胞功能障碍。本研究结果显示,OSA组NO水平较对照组有更低的表达,慢性间歇低氧条件下高血压病人血清NO水平较OSA、高血压病人更低,这与Moya等[20]研究结果一致。NO是由左旋精氨酸和氧分子结合产生的,OSA反复的低氧血症导致氧化应激使O2-生成过多,从而导致NO破坏增多,NO水平下降;而且低氧血症导致氧分子降低,NO合成减少,促进高血压的发生发展。
在氧化应激条件下,NO与O2-迅速反应形成ONOO-,从而导致3-NT的生成。研究发现内皮功能障碍时NOS表达和超氧化物产生同时增加,且与3-NT生成增加密切相关[21],而且NO有效性的减少与内皮细胞的过氧亚硝酸盐生成增加有关[6],从而加重血管内皮的损伤,本研究结果显示,各组中3-NT水平越高,NO表达水平就越低,3-NT、NO分别作为慢性间歇低氧诱发的高血压的正向、负向调节因子,相关分析发现3-NT与NO呈负相关(r=-0.524,P<0.001)。
本研究通过检测OSA合并高血压病人3-NT、NO水平,从氧化应激水平阐述OSA相关性高血压的发病机制,结果表明OSA合并高血压病人有更高水平的3-NT表达及更低水平的NO表达,而且随着OSA程度的加重,3-NT水平更高、NO水平更低,这表明OSA合并高血压病人有更高水平的氧化应激,而ONOO-清除剂逆转慢性间歇低氧诱发的高血压尚需进一步研究证实。
