含氮化合物对柴油超深度加氢脱硫催化剂运行稳定性的影响
2019-02-22葛泮珠李大东
葛泮珠, 丁 石, 张 乐, 李大东, 聂 红
(中国石化 石油化工科学研究院, 北京 100083)
由于原油劣质化程度的加剧与环保法规的日趋严格,超低硫柴油的生产成为研究的重点[1-3]。目前多数研究主要集中于高活性催化剂的开发,然而在生产超低硫柴油过程中,由于操作条件更加苛刻,催化剂的失活问题显得尤为突出。多数研究表明[4-6],原料中含氮化合物,尤其是碱性含氮化合物,能够强烈吸附在催化剂酸性位上且不易脱附进而形成焦炭,缩短了催化剂的使用寿命。但目前对长周期生产超低硫柴油过程中,不同运转阶段含氮化合物对催化剂运行稳定性的影响尚未见系统的研究报道。
因此,笔者以直馏柴油中掺炼20%的催化裂化柴油作为常用柴油,通过向常用柴油中添加一定量的含氮化合物,采用中型固定床加氢装置模拟工业运转的情况,以生产超低硫柴油为目标,考察含氮化合物对柴油超深度加氢脱硫Ni-Mo-W型催化剂运行稳定性的影响,探讨催化剂的失活原因,为预测催化剂工业运转的情况及催化剂的开发提供一定的技术指导信息。
1 实验部分
1.1 催化剂、原料油和试剂
实验所用的加氢精制催化剂为中国石化石油化工科学研究院开发并已工业应用的Ni-Mo-W/γ-Al2O3催化剂,表1为该催化剂的组成及其物理性质。

表1 Ni-Mo-W/γ-Al2O3催化剂的组成及其物理性质Table 1 Composition and physical properties of Ni-Mo-W/γ-Al2O3 catalyst
实验所用的常用柴油来自中国石化镇海炼油厂,按照常压二线柴油∶常压三线柴油∶催化裂化柴油质量比为2∶2∶1进行调配作为原料油A,其性质见表2。根据文献[7]可知,催化裂化柴油中的含氮化合物主要为喹啉、吲哚和咔唑类,由于吲哚和咔唑类含氮化合物在柴油中的溶解性较低,实验采用向原料油A中添加350 μg/g喹啉(以氮质量分数计)方式获得原料油A+BN。

表2 柴油原料油A的性质Table 2 Properties of diesel feed A
直馏煤油,采自中国石化燕山石化公司;喹啉、甲苯和环己烷,分析纯,国药集团化学试剂有限公司产品。
1.2 加氢实验
本实验选用中型固定床加氢装置,实验开始前,采用湿法对催化剂进行硫化,硫化油选用CS2质量分数为2.0%的燕山直馏煤油,在反应压力6.4 MPa、液时空速1.5 h-1、氢/油体积比300的条件下,以一定速率升温硫化;硫化结束后,采用钝化油,在反应温度320 ℃、反应压力6.4 MPa、液时空速2.0 h-1、氢/油体积比300的条件下,对催化剂进行钝化处理48 h。笔者以硫化结束后为运转时间的零点。
钝化结束后开始引入原料油,在氢分压6.4 MPa、液时空速2.0 h-1、氢/油体积比300的条件下,以控制产品硫质量分数小于10 μg/g为目标调节反应温度。分别选取钝化48 h以及运行248、448和1048 h后的催化剂为研究对象,通过对比原料油添加含氮化合物前后不同运转阶段催化剂的失活规律和失活原因,考察含氮化合物对柴油超深度加氢脱硫催化剂运行稳定性的影响。
1.3 催化剂的表征
催化剂分析表征之前,先用甲苯对催化剂进行索氏抽提,以去除催化剂表面及孔道中吸附的油分,然后在80 ℃下真空干燥4 h。采用日本HORIBA公司生产的EMIA-320V型C、S分析仪,以高温燃烧-红外检测法测定催化剂C、S元素组成。采用美国ANTEK公司的ANTEK 9000总氮分析仪,以化学发光法测定催化剂N含量。采用美国康塔仪器公司的AUTOSORB-6B 型分析仪测量催化剂样品的孔结构数据。
采用德国耐驰公司生产的STA409PC-QMS403型热重-质谱联用仪分析催化剂质量变化和逸出气体,空气流量30 mL/min,升温速率10 ℃/min,升温至800 ℃。
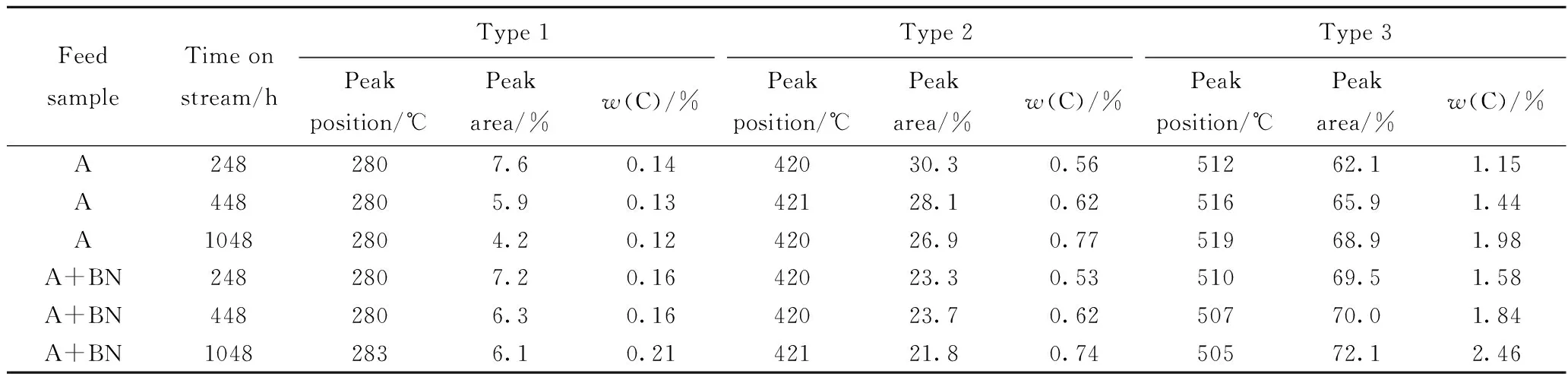
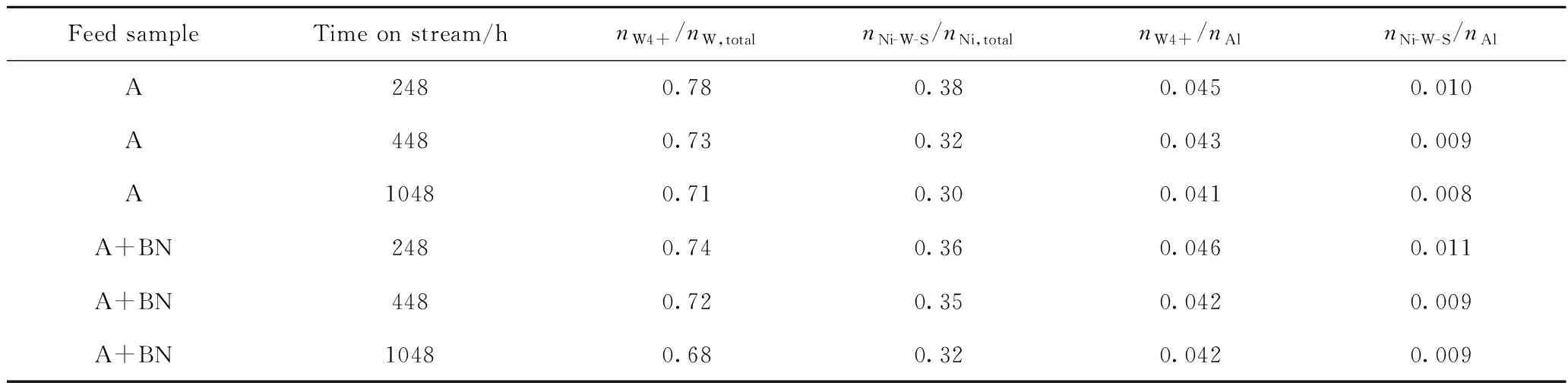
将卸出的催化剂保存在环己烷溶液中,避免接触空气,磨粉后,采用Thermo Scientific公司的ESCALab250 型X射线光电子能谱仪测定催化剂表面活性组分的化学态和种类,以及活性组分在载体上分散状态的相关信息[8-9]。激发源为非单色化的AlKαX射线,能量为1486.6 eV。分析室的基础真空约为3×10-7Pa。电子结合能用C1s峰(284.6 eV)进行能量校正。对催化剂样品的Ni2p、Mo3d和W4f谱图进行解析,由于催化剂中Mo的含量较低,各催化剂的Mo硫化度相差不大,主要对W4f和Ni2p谱图进行解析,可获得硫化态加氢催化剂的W硫化度(nW4+/nW,total)和Ni-W-S相比例(nNi-W-S/nNi,total),具体解析方法见文献[10-11]。对于本实验Ni-Mo-W型催化剂,由于加氢反应主要在Ni-W-S活性相上进行,因此Ni-W-S相的比例可在一定程度上反映活性中心数目的变化情况。同时利用XPS得到的元素相对含量计算负载金属在载体表面的分散状况,笔者以nW4+/nAl与nNi-W-S/nAl表示活性金属的分散度。

(1)
(2)
其中,Li和Ni分别为第i个晶粒的长度和层数;n′为统计区域内WS2晶粒的总数量。
由于加氢反应主要在活性相的边缘活性位上进行,用边缘W含量占总W含量的比值fW反映活性中心数目的变化情况,通常按式(3)和式(4)计算fW[12-14]。
(3)
(4)
其中,nW,edge为边缘位的W原子数量;nW,total为总的W原子数量;nW,i为片晶i的边缘位上W原子数量。
2 结果与讨论
2.1 Ni-Mo-W型催化剂的失活规律
对于柴油加氢脱硫反应的表观反应级数,有不少学者进行了大量的研究,普遍认为中间馏分油在缓和加氢脱硫及深度加氢脱硫时的表观反应级数在1.0~2.0之间[15-17]。由于在实际操作过程中,存在提温滞后的问题,为更准确地分析催化剂的失活速率,根据文献调研拟采用反应级数为1.2,活化能Ea=80 kJ/mol[18],目标产品硫质量分数为9 μg/g,对实际反应温度进行归一化处理。图1为柴油原料油添加含氮化合物前后Ni-Mo-W型催化剂运行稳定性的变化情况。根据图1中反应温度的提温情况,将运转时间48~248 h阶段定义为运转初期阶段,该阶段催化剂的活性定义为催化剂的初活性;将运转时间248~448 h阶段定义为运转中前期阶段;将运转时间448~1048 h阶段定义为运转中期阶段。图1中每个阶段所标的数据为该阶段催化剂的失活速率情况。

图1 含氮化合物对深度加氢脱硫Ni-Mo-W型催化剂运行稳定性的影响Fig.1 Effect of nitrogen compounds on the running stability ofultra-deep hydro-desulfurization catalyst operation A; A+BNReaction conditions: p=6.4 MPa; LHSV=2.0 h-1;V(H2)/V(Oil)=300
由图1可知,原料油添加含氮化合物前后,Ni-Mo-W 型催化剂的失活规律基本类似,均是在运转初期催化剂反应活性损失最为明显,之后催化剂从初期的快速失活阶段逐渐过渡到中期缓慢失活阶段。相同运转时间,原料油添加含氮化合物后,与加工常用柴油(即原料油A)相比,在运转初期(48~248 h)阶段,反应温度提高7~10 ℃,说明原料油添加含氮化合物明显影响了Ni-Mo-W型催化剂的初活性。
根据上述分析可知,原料油添加含氮化合物后只是影响了Ni-Mo-W型催化剂的初活性,而对催化剂的稳定性影响并不大。这可能是由于含氮化合物有较强的竞争吸附作用,原料油中添加含氮化合物后对加氢脱硫反应产生强烈的抑制作用,从而使催化剂的初活性大幅下降[19-20],但是在反应初期之后,具有强吸附能力的活性中心已被占据,吸附能力减弱,使得含氮化合物在反应中期竞争吸附作用表现不明显,因此对催化剂的稳定性影响不大。
2.2 Ni-Mo-W型催化剂的表征结果
2.2.1 催化剂的C、S、N元素分析
对原料油添加含氮化合物前后不同运转时间卸出的Ni-Mo-W型催化剂进行抽提处理,然后进行催化剂C、S、N元素分析,结果如表3所示。
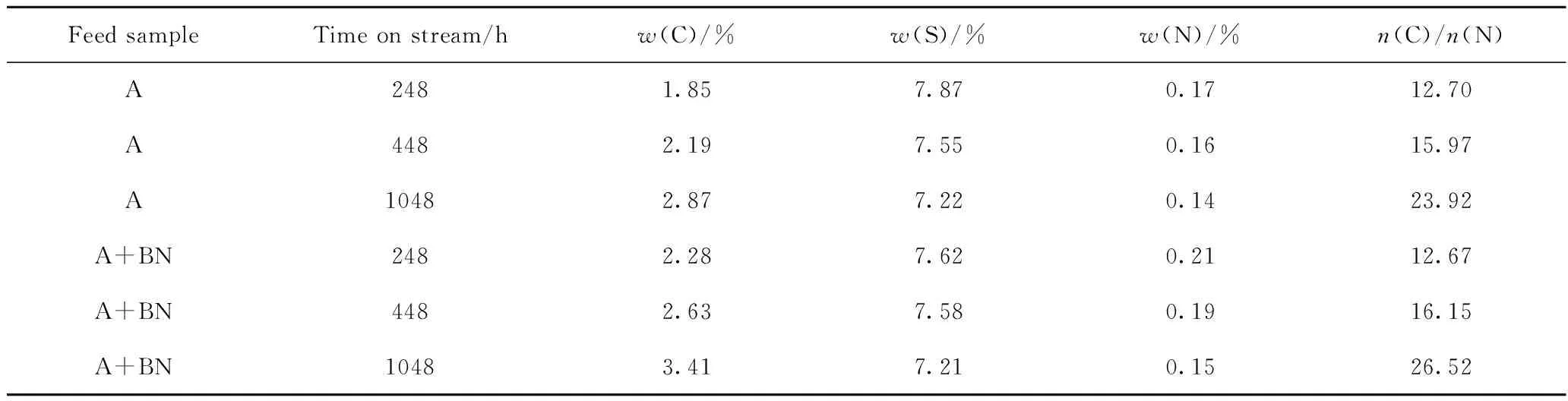
表3 卸出的深度加氢脱硫Ni-Mo-W型催化剂的C、S、N元素分析结果Table 3 Analytical results of C, S, N contents in the discharged ultra-deep hydro-desulfurization catalyst
根据表3可知,原料油中添加含氮化合物前后,随着运转时间的延长,Ni-Mo-W型催化剂的积炭量均逐渐增加。相同运转时间条件下,对比原料油添加含氮化合物前后催化剂积炭量的变化发现,在运转初期248 h,原料油添加含氮化合物后,催化剂积炭量增加了0.43百分点,之后积炭量的增加幅度变化并不大。说明原料油添加含氮化合物后主要增加了运转初期催化剂积炭的形成,在运转初期之后对催化剂积炭量的影响并不大。根据S、N含量的变化情况可知,原料油添加含氮化合物后,相同运转时间,S含量的变化并不明显,N含量略有提高,C/N原子比略有提高。Dong等[21]研究认为,原料油中添加含氮化合物后,催化剂上N含量的增加可能是催化剂上吸附的未反应的含氮化合物,也有可能是含氮化合物的聚合物。
原料油添加含氮化合物前后反应温度与Ni-Mo-W型催化剂的积炭量随运转时间的变化关系如图2所示。由图2可知,原料油添加含氮化合物前后,催化剂反应温度的变化与积炭量的变化均呈现出较好的相关性。说明含氮化合物主要是通过竞争吸附的作用影响了Ni-Mo-W型催化剂的初活性和初期积炭量,而在运转初期之后,催化剂反应温度与积炭量的变化同样呈现出较好的相关性,两者均随运转时间的延长逐渐趋于平缓。
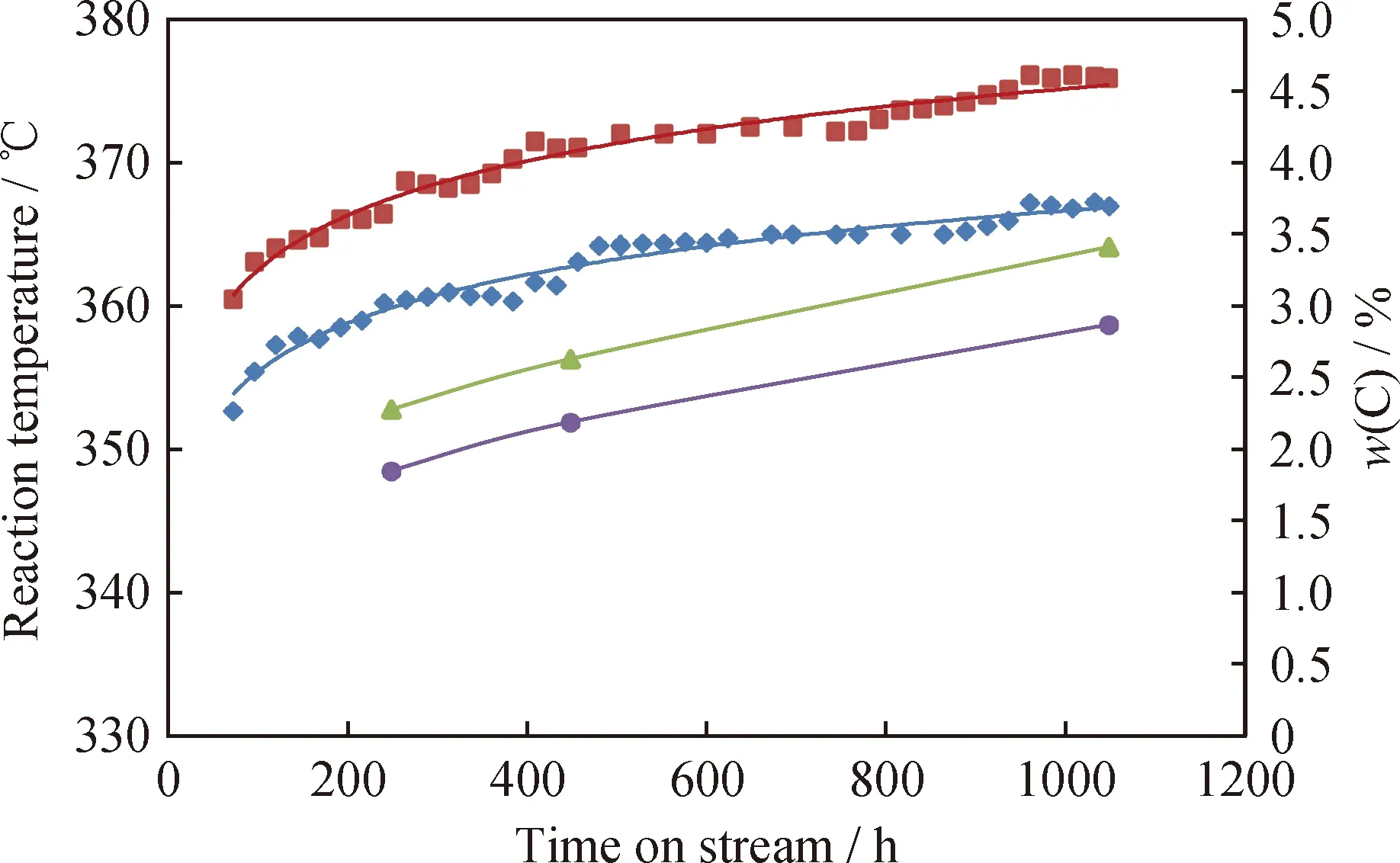
图2 深度加氢脱硫Ni-Mo-W型催化剂>反应活性与积炭量的关联Fig.2 Relationship between ultra-deep hydro-desulfurizationcatalyst activity and carbon deposit Reaction temperature, A; Reaction temperature, A+BN; Carbon deposition, A; Carbon deposition, A+BNReaction conditions: p=6.4 MPa; LHSV=2.0 h-1;V(H2)/V(Oil)=300
2.2.2 催化剂的N2吸附-脱附表征
通过N2吸附-脱附的表征研究原料油添加含氮化合物前后对Ni-Mo-W型催化剂孔结构的影响,表4为原料油添加含氮化合物前后不同运转时间 Ni-Mo-W 型催化剂的比表面积、孔体积的变化情况。

表4 不同运转时间下深度加氢脱硫Ni-Mo-W型催化剂的比表面积(SBET)和孔体积(Vp)Table 4 Specific surface area (SBET) and pore volume (Vp) ofultra-deep hydro-desulfurization catalystwith different time on stream
由表4可知,原料油添加含氮化合物前后,随着运转时间的延长,Ni-Mo-W型催化剂的比表面积和孔体积均呈现逐渐降低的趋势。相同运转时间条件下,原料油添加含氮化合物后,催化剂的比表面积和孔体积略有下降,这可能是由于原料油添加含氮化合物后,反应温度的提高导致催化剂积炭量增加,积炭的形成覆盖了催化剂的一些比表面积,堵塞了催化剂的部分孔道,导致催化剂比表面积和孔体积略有下降[22]。
原料油添加含氮化合物前后Ni-Mo-W型催化剂孔径分布的变化情况如图3所示。由图3可知,原料油添加含氮化合物前后,随着运转时间的延长,催化剂的最可几孔径基本未变。相同运转时间条件下,原料油添加含氮化合物前后催化剂孔体积分布变化幅度较小,说明在常用柴油原料中添加含氮化合物对催化剂孔结构的影响较小。
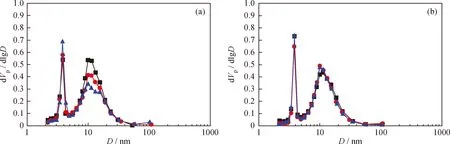
图3 深度加氢脱硫Ni-Mo-W型催化剂孔体积分布曲线Fig.3 Ultra-deep hydro-desulfurization catalyst pore volume distribution curves(a) A; (b) A+BNTime on stream/h: 248; 448; 1048
2.2.3 催化剂的TG-MS表征
原料油添加含氮化合物前后不同运转时间Ni-Mo-W 型催化剂的热重(TG)及质量损失速率(DTG)曲线如图4所示,催化剂的质谱(MS)谱图如图5所示。
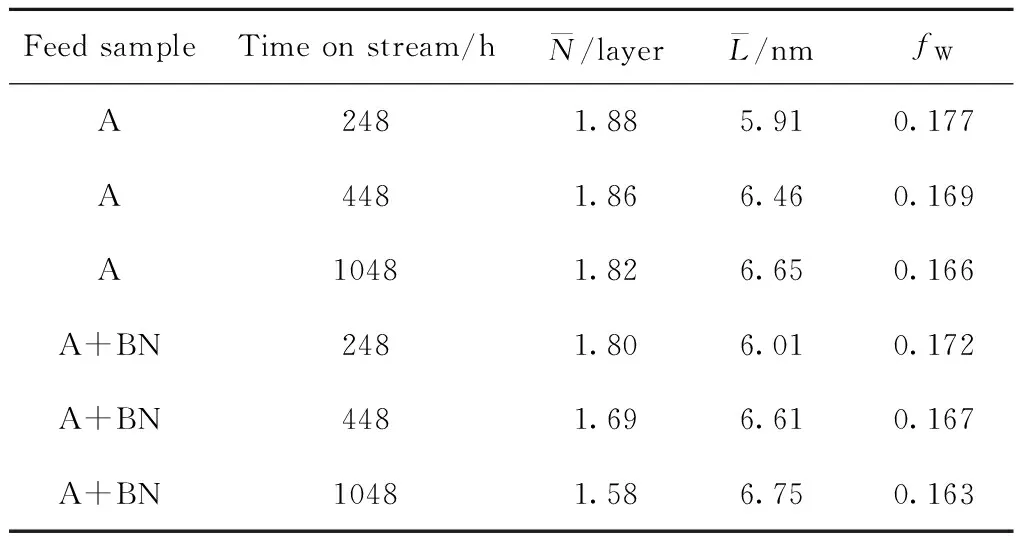
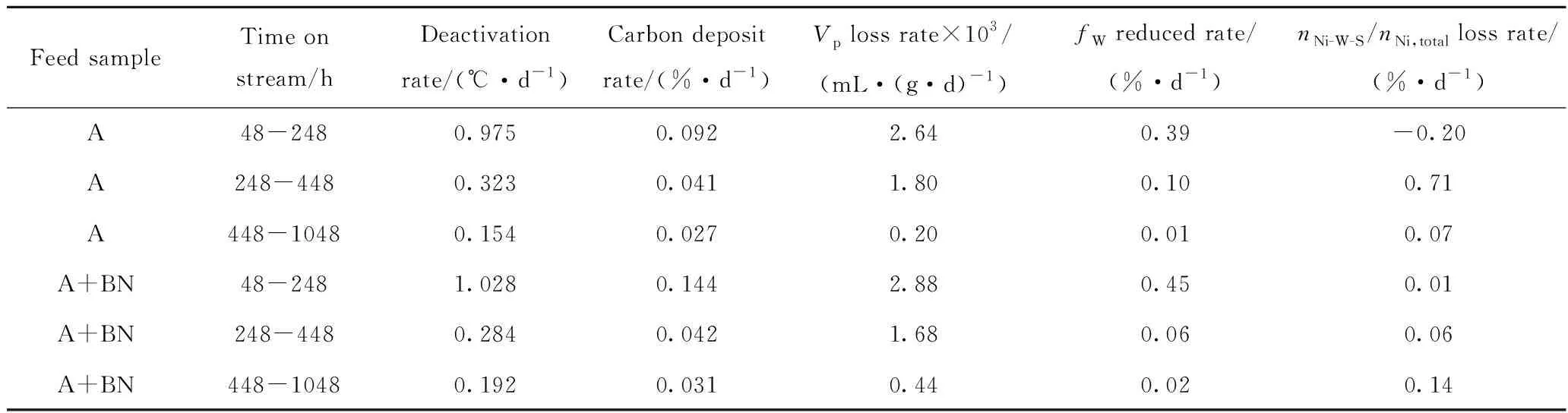
根据图4可知,原料油添加含氮化合物前后,不同运转时间卸出Ni-Mo-W型催化剂的TG曲线均存在3个明显区域[23-24]:第一区域为起始温度 根据图5可知,相同运转时间条件下,原料油添加含氮化合物前后同种物质的谱峰位置相差不大,说明原料油添加含氮化合物后对Ni-Mo-W型催化剂积炭的类型基本无影响。其中H2O的谱峰主要在小于200 ℃的低温区域出峰,该处谱峰主要是由于催化剂表面吸附的少量水挥发形成。对比CO2与NO2的谱峰可知,2种物质的谱峰位置非常相似,说明催化剂积炭中含有部分氮化物;而对比CO2与SO2谱峰可知,两者强弱谱峰的出峰位置正好相反,说明不同类型积炭中C、S含量的分布不同。每种物质的具体谱峰信息见文献[25]。为进一步分析原料油添加含氮化合物后不同类型积炭量的变化情况,对不同运转时间的CO2谱峰进行分峰处理,计算每种类型积炭谱峰的峰位、峰面积以及积炭量,具体数据如表5所示。 图4 不同运转时间下卸出的深度加氢脱硫Ni-Mo-W型催化剂的TG曲线Fig.4 TG curves for the discharged ultra-deep hydro-desulfurization catalyst with different time on stream(a) A; (b) A+BNTime on stream/h: 248; 448; 1048 图5 不同运转时间下卸出的深度加氢脱硫Ni-Mo-W型催化剂的质谱图Fig.5 Mass spectra fitting curves of the discharged ultra-deep hydro-desulfurization catalyst with different time on stream(a) H2O; (b) CO2; (c) SO2; (d) NO2 248 h-A; 448 h-A; 1048 h-A; 248 h-A+BN; 448 h-A+BN; 1048 h-A+BN 根据表5中CO2谱峰的分峰情况可知,Ni-Mo-W型催化剂上的积炭可分为3种类型:类型1积炭(280 ℃左右)、类型2积炭(420 ℃左右)、类型3积炭(510 ℃左右)。原料油添加含氮化合物前后,相同运转时间条件下,类型1和类型2积炭的谱峰位置基本不变,而类型3高温型积炭的峰温向低温区移动。根据表5中不同类型谱峰的峰面积和积炭量的变化可知,原料添加含氮化合物前后,随着运行时间的延长,类型1积炭和类型2积炭的峰面积比例和积炭量变化不大,但类型3积炭的峰面积比例和积炭量均逐渐增加。这说明原料油添加含氮化合物后,与活性相相关积炭的增加幅度较小,主要促进了类型3高温型积炭的形成。以运转1048 h后的Ni-Mo-W型催化剂为例,原料油添加含氮化合物前后催化剂总积炭量增加了0.54百分点,其中类型3积炭量增加了0.48百分点,两者数据相差不大,说明原料油添加含氮化合物后Ni-Mo-W型催化剂上增加的积炭主要以类型3高温型积炭为主。 表5 不同运转时间下卸出的深度加氢脱硫Ni-Mo-W型催化剂CO2各类型谱峰的比例Table 5 CO2 peaks position and proportion of the discharged ultra-deep hydro-desulfurization catalyst with different time on stream 为进一步分析Ni-Mo-W型催化剂的失活原因与积炭类型的关系,将原料油添加含氮化合物前后反应温度的变化与催化剂上不同类型积炭量的变化进行关联,结果如图6所示。 图6 不同运转时间下卸出的深度加氢脱硫Ni-Mo-W型催化剂积炭类型与反应活性的关联Fig.6 Relationship of the discharged ultra-deep hydro-desulfurization catalyst carbon species andreactivity with different time on stream(a) A; (b) A+BN Type 1; Type 2; Type 3; Reaction temperatureReaction conditions: p=6.4 MPa; LHSV=2.0 h-1; V(H2)/V(Oil)=300 根据图6可知,原料油添加含氮化合物前后Ni-Mo-W型催化剂活性的损失与类型3高温型积炭的形成均存在明显的相关性。这可能主要是由于类型3高温型积炭的形成,导致部分Ni-W(Mo)-S活性相结构边缘被积炭覆盖,限制了较大的分子在活性中心上的反应,因此导致部分活性中心得不到充分利用,最终导致催化剂活性的下降[19,26-27]。 2.2.4 催化剂的XPS表征 原料油添加含氮化合物前后不同运转时间条件下卸出Ni-Mo-W型催化剂的XPS表征结果如表6所示。 由表6可知,原料油添加含氮化合物前后,随着运转时间的延长,Ni-Mo-W型催化剂的W硫化度、Ni-W-S相的比例和活性金属的分散度均逐渐下降;相同运转时间下,与未添加含氮化合物的原料油A相比,原料油添加含氮化合物后,催化剂的W硫化度、Ni-W-S相的比例和活性金属的分散度相差不大,说明原料油添加含氮化合物后对Ni-Mo-W型催化剂的W硫化度、Ni-W-S相的比例和活性金属的分散度影响并不明显。 表6 不同运转时间下卸出的深度加氢脱硫Ni-Mo-W型催化剂的XPS表征结果Table 6 XPS characterization results of the discharged ultra-deep hydro-desulfurization catalyst with different time on stream 2.2.5 催化剂的TEM表征 表7 不同运转时间下卸出的深度加氢脱硫Ni-Mo-W型催化剂上可观察到的WS2条纹统计结果Table 7 Statistical results of observed WS2 stripes on thedischarged ultra-deep hydro-desulfurization catalystwith different time on stream 由表7可知,随着运转时间的延长,无论原料油中是否添加含氮化合物,Ni-Mo-W型催化剂上WS2多层片晶的平均堆叠层数均逐渐降低,片晶的平均长度均逐渐增加,边缘W的比例fW值均逐渐降低。相同运转时间下,与未添加含氮化物的原料油A相比,原料油添加含氮化合物后,Ni-Mo-W型催化剂上WS2多层片晶的平均堆叠层数、片晶的平均长度和边缘W的比例fW值变化并不明显,说明原料油添加含氮化合物后对Ni-Mo-W型催化剂上活性相的结构影响并不大。 结合前面的分析,对原料油添加含氮化合物前后Ni-Mo-W型催化剂失活规律和失活原因进行了系统的总结,统计了不同运转阶段催化剂的失活速率与积炭速率、孔体积损失速率、边缘W比例的减少速率和Ni-W-S相比例的损失速率的变化情况,具体数据如表8所示。 表8 不同运转时间下卸出的深度加氢脱硫Ni-Mo-W型催化剂失活原因分析Table 8 Reasons of the discharged ultra-deep hydro-desulfurization catalyst deactivation with different time on stream 根据表8可知,原料油添加含氮化合物前后,随运转时间的增加,Ni-Mo-W型催化剂的积炭速率、孔体积损失速率、边缘W比例减少速率与催化剂失活速率的变化规律均基本一致,而Ni-W-S相比例损失速率与催化剂失活速率的变化规律相差较大,说明原料油添加含氮化合物前后Ni-Mo-W型催化剂活性的损失均与积炭的形成、孔体积的损失和边缘W比例的下降密切相关。这可能是由于积炭的形成,导致催化剂的孔体积下降,催化剂活性中心的可接近性降低,同时在反应过程中活性相结构的变化导致边缘W比例的降低,多方面的共同作用导致催化剂活性的下降。而原料油添加含氮化合物后Ni-Mo-W型催化剂初活性的损失,可能主要是由于含氮化合物的竞争吸附作用导致加氢活性的降低。 (1)与加工常用柴油相比,原料油添加含氮化合物后,主要影响了Ni-Mo-W型催化剂的初活性,运转初期反应温度提高了7~10 ℃,但对催化剂的稳定性影响不大,均是在运转初期反应活性损失最为明显,之后从初期的快速失活阶段逐渐过渡到中期缓慢失活阶段。 (2)根据催化剂失活原因的分析,原料油添加含氮化合物前后Ni-Mo-W型催化剂失活的主要原因基本相一致,均与表面积炭的形成、孔体积的损失和边缘W比例的下降密切相关。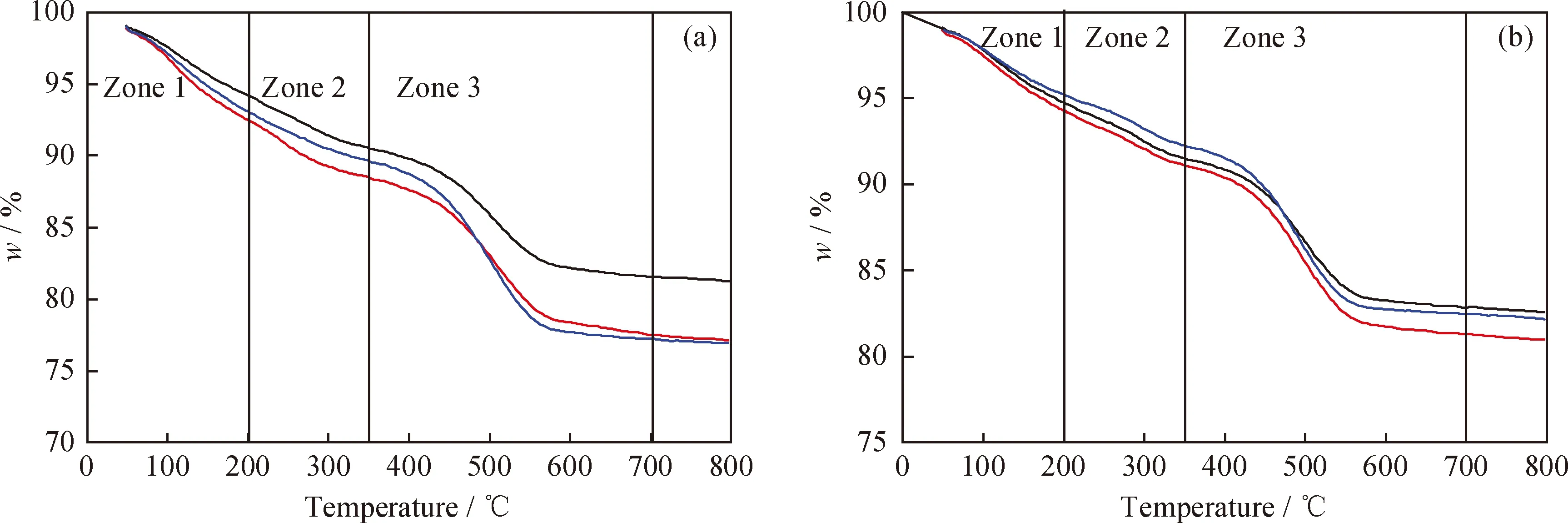


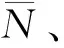
2.3 Ni-Mo-W型催化剂失活原因分析
3 结 论
