正相柱色谱法分离制备陈皮中橘皮素和川陈皮素
2019-02-14吕小健徐兰英李士明王运利
吕小健,许 引,徐兰英,李士明,龙 涛*,王运利
(1.武汉纺织大学 化学与化工学院,湖北 武汉 430200;2.黄冈师范学院 化学化工学院 催化材料制备及应用湖北省重点实验室,湖北 黄冈 438000)
陈皮又称橘皮、广陈皮,为芸香科植物橘及其栽培变种的成熟果皮,是一种药食同源的中药,有理气调中,燥湿化痰功效[1-3]。已有研究表明,陈皮中特有的黄酮类化合物具有抗氧化[4-6]、抗炎[7-9]、抗癌[10-12]、抗菌[13]、保肝作用[14]、保护心脑血管系统[15]等生理活性。橘皮素和川陈皮素均为多甲氧基黄酮类化合物,属于陈皮中的特有黄酮,对陈皮的生物活性起到了重要作用,其化学结构式见图1。
川陈皮素能通过抑制肝脏脂肪酸生物合成和增加脂肪酸氧化,来防止肝脏脂肪变性和血脂异常,具有预防肥胖、辅助降血脂的作用[16-18]。橘皮素能通过诱导CYP1A1/CYP1B1介导的代谢途径,可抑制乳腺癌细胞的增殖[19]。随着陈皮黄酮研究的不断深入,许多生物活性研究尤其是动物实验受到单体数量的制约,因此亟需一种简便、高效的大量分离制备陈皮中橘皮素和川陈皮素的方法。

图1 橘皮素(a)和川陈皮素(b)的结构式Fig.1 Structural formulas of the tangeretin(a)and nobiletin(b)
本研究采用柱色谱法同时分离制备纯度较高的橘皮素和川陈皮素,采用柱层析硅胶为分离介质,不同比例的乙酸乙酯-石油醚混合液为洗脱溶剂,同时分离制备较高纯度的橘皮素和川陈皮素。该方法成本低,简单易行,可以同时大量制备橘皮素和川陈皮素,以期为陈皮生物活性的深入研究提供了重要的物质基础。
1 材料与方法
1.1 材料与试剂
陈皮:市售。
无水乙醇、石油醚、乙酸乙酯、正己烷、二氯甲烷(均为分析纯),甲醇、乙腈(均为色谱纯):国药集团上海化学试剂有限公司。橘皮素和川陈皮素对照品:本实验室自制(经过1H-NMR、13C-NMR和ESI-MS鉴定结构,纯度采用HPLC峰面积归一化法计算均≥98%)。
1.2 仪器与设备
GWX-9623MB型电热鼓风干燥箱:上海博迅实业有限公司医疗设备厂;2XZ-1型旋片式真空泵:临海市谭氏真空设备有限公司;CCA-20型低温冷却水循环泵:巩义市予华仪器有限责任公司;RE-52AA型旋转蒸发仪:上海亚荣生化仪器厂;ZF7型三用紫外分析仪:上海舜宇恒平科学仪器有限公司;BSA124S分析天平:赛多利斯科学仪器(北京)有限公司;1260型高效液相色谱仪(high performance liquid chromatography,HPLC):美国安捷伦公司;UltiMate 3000 plus型液相色谱-质谱(liquid chromatography-massspectrometry,HPLC-MS)联用仪:美国Thermo Fisher公司。
1.3 实验方法
1.3.1 色谱条件
HPLC 条件:Agilent C18(4.6 mm×150 mm,5 μm)液相色谱柱;流动相:乙腈-水(梯度:0~15 min,40%~50%乙腈;15~20 min,50%~70%乙腈;20~25 min,70%~85%乙腈;25~30 min,85%~40%乙腈);流速:1.0 mL/min;柱温:35℃;检测波长:326 nm;进样量:10μL。
质谱条件:电子电离(electrospray ionization,EI)源;检测方式为正离子模式,鞘气流速34.5 kPa;喷雾电压75 kV;毛细管温度320℃;辅助气加热器温度300℃。
1.3.2 对照品储备液的制备
分别精密称取5.0 mg川陈皮素和橘皮素对照品,用色谱甲醇超声辅助溶解并定容至10 mL容量瓶中,配制成0.5 mg/mL对照品储备液,保存于4℃冰箱中,备用。
1.3.3 橘皮素和川陈皮素样品的提取分离及检测
乙醇浸提:称取干陈皮粉,用无水乙醇于室温条件下浸提48 h,料液比1∶10(g∶mL),过滤并收集滤液,将滤液置于旋转蒸发仪减压蒸馏得陈皮粗提物Ⅰ。
有机溶剂萃取:将陈皮粗提物Ⅰ用适量蒸馏水配成混合悬浮液,混匀后加入一定体积的正己烷萃取2~3次,再用二氯甲烷萃取2~3次,合并二氯甲烷层,减压浓缩除去二氯甲烷溶剂即得陈皮粗提物Ⅱ。
装柱:称取200~300目的硅胶70 g置于烧杯中,加入100 mL石油醚(即加入干硅胶体积1倍的溶剂),用玻璃棒充分搅拌,搅成匀浆。将4 cm×30 cm层析柱管清洗干净并保持干燥,垂直固定在铁架台上,取一小团脱脂棉用溶剂润湿后塞入管底。将硅胶均匀的装入层析柱中,保持上部水平。然后在硅胶柱顶部加入适量海砂,用胶棒轻轻敲打海沙表面至水平。
洗脱:将适量陈皮黄酮混合物溶于少量二氯甲烷得悬浊液。湿法上样后,依次用不同比例的乙酸乙酯-石油醚溶液进行洗脱,乙酸乙酯的体积分数分别为20%、30%、40%、50%、60%,每40 mL收集一瓶,从第一瓶按照顺序编号,直到60%洗脱剂洗脱完毕。
检测:分别将不同编号样品收集瓶中的溶剂旋转蒸发至干,得到的固体物质用色谱甲醇溶解,过0.22μm滤膜后,直接注入进样瓶以供HPLC分析。
2 结果与分析
2.1 橘皮素和川陈皮素标准曲线
将对照品储备液采用逐步稀释法稀释至2.5μg/mL、50μg/mL、100μg/mL、125μg/mL、250μg/mL、500μg/mL,得不同质量浓度的系列标准工作溶液,按1.3.1中色谱条件测定,记录峰面积。以每个标准工作溶液对应的峰面积(y)和其质量浓度(x)进行线性回归分析,得到川陈皮素标准曲线回归方程:y=31.719x+89.264(R2=0.999 9);橘皮素标准曲线回归方程:y=37.632x+10.145(R2=0.999 8)。结果显示,川陈皮素和橘皮素在2.5~500μg/mL范围内线性关系良好。
2.2 陈皮粗提物Ⅱ中川陈皮素、橘皮素含量的检测
准确称取5 mg陈皮粗提物Ⅱ用色谱甲醇定容至10 mL,取1 mL溶液过0.22μm滤膜后经HPLC分离,检测得到化合物的峰面积,代入标准曲线回归方程计算,得陈皮黄酮混合物中川陈皮素含量为1.6 mg,橘皮素含量为1.1 mg。
2.3 陈皮粗提物Ⅰ与陈皮粗提物Ⅱ成分比较
川陈皮素和橘皮素标准品及二氯甲烷萃取陈皮粗提物前后经HPLC检测比较其中物质变化,结果如图2所示。
由图2A可知,标准品川陈皮素和橘皮素的保留时间分别为13.1 min、19.3 min。对比图2B和图2C可知,二者均含有较多川陈皮素和橘皮素,可见这两种多甲氧基黄酮是陈皮中的主要黄酮。陈皮粗提物Ⅰ经有机溶剂萃取后,多甲氧基黄酮纯度明显提高。这是因为正己烷萃取除去了稠膏中的非极性物质,且多甲氧基黄酮在二氯甲烷中溶解度很好,几乎所有的多甲氧基黄酮都溶于二氯甲烷层,而极性较强的杂质则大部分留在水溶液中。因此,陈皮粗提物Ⅰ通过正己烷和二氯甲烷萃取之后,混合组分得到了初步纯化,为后续的柱色谱分离得到陈皮多甲氧基黄酮单体奠定了基础。

图2 对照品(A)、陈皮粗提物Ⅰ(B)、陈皮粗提物Ⅱ(C)的高效液相色谱图Fig.2 HPLC chromatogram of reference substance(A),crude extractsⅠ(B)and crude extractsⅡ(C)from tangerine peel
2.4 正相柱色谱法分离陈皮黄酮混合物
按照1.3.3方法装柱,二氯甲烷萃取得到的陈皮粗提物Ⅱ上样后,依次用不同比例的乙酸乙酯-石油醚溶液进行洗脱,分别收集到25个样品瓶中,1~5#、6~10#、11~15#、16~20#、21~25#样品瓶分别是质量分数为 20%、30%、40%、50%、60%乙酸乙酯的洗脱剂洗脱所得溶液,同时用薄层色谱(thin layerchromatography,TLC)及HPLC跟踪监测。HPLC检测结果中,体积分数为30%乙酸乙酯洗脱后,流出物中开始出现川陈皮素和橘皮素,其中第8号样品瓶检测结果见图3。由图3可知,在保留时间21.9 min处有一个较大的物质峰,但在保留时间19.3 min处出现了一个较小的色谱峰,通过保留时间、紫外光谱及标准加入法确证了保留时间为19.3 min的物质为橘皮素。结果说明,体积分数为30%的乙酸乙酯极性较弱,根据相似相溶原理,可将陈皮黄酮混合物中极性较弱的物质洗脱下来,但在此极性条件下,橘皮素已经开始被淋洗下来,但含量很低,因而可以加大淋洗液极性继续进行洗脱。

图3 30%乙酸乙酯-70%石油醚洗脱剂洗脱样品8#的色谱图Fig.3 Chromatogram of the eluant of sample 8#eluted by 30%ethyl acetate and 70%petroleum ether
用体积分数40%的乙酸乙酯洗脱剂洗脱,主要得到一种物质,其中第12号管的液相色谱分离图如图4(a),通过面积归一化法进行计算,其纯度达到98.1%。与橘皮素的标准品比对发现,其保留时间和紫外吸收一致。取该样品同时进行MS检测,结果见图4(b),其分子离子峰为372.7,和文献报道一致[20],进一步证实分离得到的物质为橘皮素。

图4 12#样品的色谱图(a)及质谱图(b)Fig.4 Chromatogram(a)and mass spectrum(b)of sample 12#
用50%乙酸乙酯洗脱之后主要得到4种物质,其中第16、18#管的色谱分离结果见图5。由图5可知,增大洗脱剂浓度,洗脱液中还残留橘皮素,但明显看到极性较大的物质开始被洗脱,保留时间为13.1 min时出现了目标色谱峰,根据保留时间和紫外光谱定性初步确定其为川陈皮素。18号管中橘皮素色谱峰继续减小,川陈皮素色谱峰明显增加。表明在此洗脱剂条件下,川陈皮素逐渐被洗脱,要想洗脱得到纯度较高的川陈皮素,则需要继续增大洗脱剂浓度。
用体积分数60%的乙酸乙酯洗脱剂洗脱,取第21号样品进行检测,结果如图6所示。由图6(a)可知,图6(a)中只出现了一个明显的色谱峰,通过保留时间和紫外光谱定性,初步确定该物质为川陈皮素。通过峰面积归一化法进行计算,其纯度达98.6%。取该样品同时进行MS检测,结果见图6(b),其分子离子峰为402.4,和文献报道一致[20],进一步证实分离得到的物质为川陈皮素。
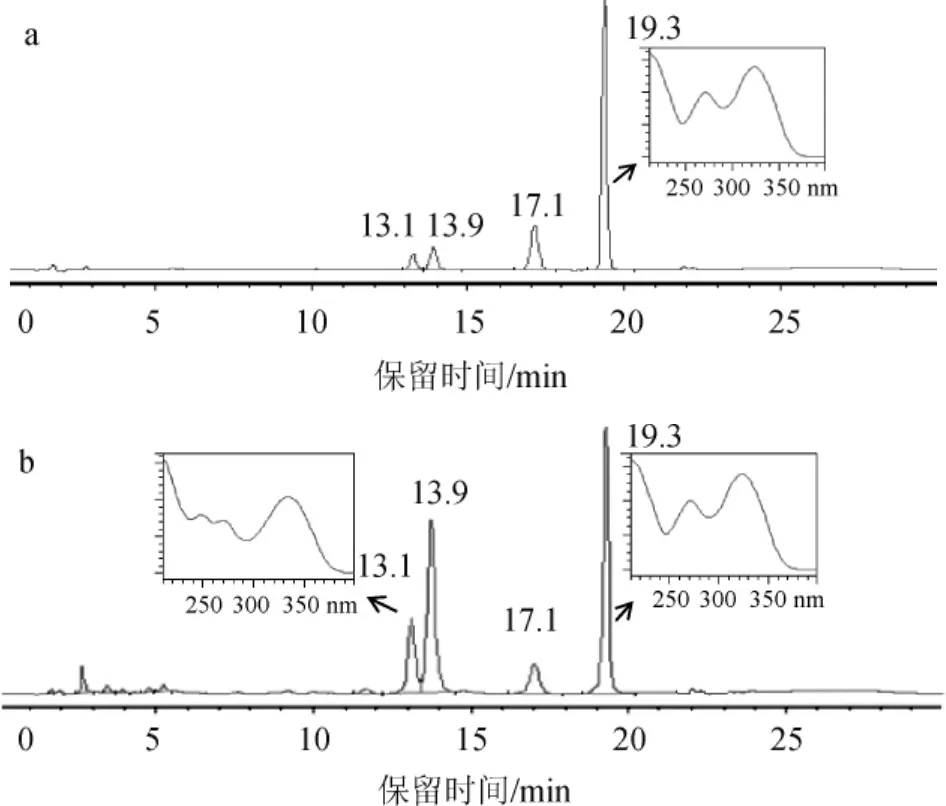
图5 50%乙酸乙酯-50%石油醚洗脱剂洗脱样品16#(a)和18#(b)的色谱图Fig.5 Chromatogram of the eluant of sample 16#(a)and 18#(b)eluted by 50%ethyl acetate and 50%petroleum ether
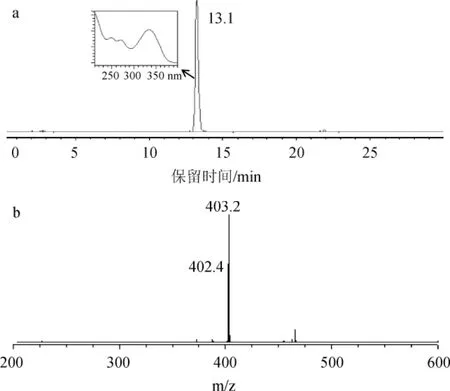
图6 21#样品的色谱图(a)及质谱图(b)Fig.6 Chromatogram(a)and mass spectrum(b)of sample 21#
3 结论
采用醇提结合硅胶柱色谱分离方法,以乙酸乙酯-石油醚溶液为洗脱剂进行洗脱,可以从陈皮中同时分离制备得到高纯度的橘皮素和川陈皮素单品,经HPLC进行纯度分析,并经质谱联用对其结构进行了确证。该方法的确立,为陈皮黄酮的生物活性筛选和体内实验的深入研究奠定了物质基础。
