以降解产物的量表征西罗莫司口服溶液质量的探讨
2019-01-30赵敬丹闻宏亮秦峰刘浩
赵敬丹 闻宏亮 秦峰 刘浩
(上海市食品药品检验所,上海 201203)
西罗莫司(sirolimus)又称雷帕霉素(rapamycin),是1975年发现的亲脂性大环内酯类免疫抑制剂[1]。1999年10月惠氏制药研制的西罗莫司口服溶液在美国首次上市,2005年国内有3家企业获批生产,主要适用于接受肾移植的患者,预防器官排斥。现行各国药典均未收载西罗莫司及其口服溶液,执行标准均为国家食品药品监督管理总局标准[2-4]。
西罗莫司口服溶液中的辅料主要有吐温80及Phosal 50PG等。其中,Phosal 50PG是主要组成部分,其性状为黏稠液体,是复合改性磷脂,主要包括磷脂酰胆碱、丙二醇、单甘油脂肪酸酯、乙醇、大豆脂肪酸和L-抗坏血酸棕榈酸酯。经本试验证实:吐温80和Phosal 50PG在西罗莫司紫外吸收光谱范围内均有吸收,这增加了采用紫外检测器测定西罗莫司口服溶液中有关物质的难度。
由于辅料的干扰,现行标准分别采用质谱法或通过改变检测波长控制其有关物质。其中,质谱法采用选择性离子监测的方式控制开环降解物、异构体和同系物的量,本试验发现:该方法重现性差,通用性差。而液相色谱法仅控制西罗莫司发酵工艺中产生的杂质—去甲基西罗莫司,不控制降解产物,并且存在明显的辅料干扰现象,实验结果重复性较差。
本文首先采用自建的西罗莫司有关物质分析方法[5],但试验结果显示,辅料的色谱保留“贯穿”整个色谱分析过程。由于Phosal50PG具有类似蜂蜜一样的黏稠度,在分析过程中不可避免会干扰测定。为了解决该问题,分别尝试利用溶解性的差异沉淀辅料或固相萃取除去辅料的方法及正相色谱法等,但沉淀后辅料仍干扰西罗莫司工艺杂质及开环降解杂质的测定;固相萃取法操作繁琐,需兼顾提取回收率,且样品处理后部分辅料仍干扰先于西罗莫司色谱峰洗脱的工艺杂质的定量测定;正相色谱法存在多次进样后,色谱柱柱效明显下降的现象。因此,为了有效地评价西罗莫司口服溶液的质量,有必要建立通过控制特定降解产物的量评价产品质量的分析方法。
本研究发现:西罗莫司在酸、碱、加热等条件下均不稳定,在其相对保留时间约为0.6处均有一个较明显的降解产物色谱峰,经质谱分析及对照品验证,确认该色谱峰为断雷帕霉素。稳定性试验表明,断雷帕霉素是西罗莫司主要的开环降解杂质,并且其量的增加趋势和西罗莫司含量的降低呈相关性。因此,本文采用通过控制降解杂质—断雷帕霉素量的方式间接表征西罗莫司口服溶液的质量。新建的方法专属性好,重现性好,通用性强,为完善质量标准,有效监控产品质量奠定了基础。
1 仪器与试药
1200型高效液相色谱仪(Agilent公司,配备二极管阵列检测器);2695型高效液相色谱仪(Waters公司,配备二极管阵列检测器);LC-20AD型高效液相色谱仪(Shimadzu公司,配备紫外检测器)。CP225D型电子天平(德国Sartorius公司)。乙腈,叔丁基甲醚均为色谱纯,甲酸铵,甲酸及其他试剂均为分析纯,水为Milli-Q超纯水。西罗莫司对照品(浙江省食品药品检验研究院,批号:20151112,纯度:99.7%,图1);断雷帕霉素(辉瑞制药有限公司,批号:L20715-182-A,纯度:80%,图1)。西罗莫司口服溶液(A企业,批号160101,规格50mL:50mg;B企业,批号150916,规格50mL:50mg;C企业,批号FKC2602001,规格20mL:20mg;均为国内抽样样品)。
2 方法与结果
2.1 色谱条件与系统适用性试验

图1 对照品结构式Fig.1 Structures of standard substances
色谱柱:Kromasil100-5 C18(4.6mm×250mm,5μm);检测波长为277nm;流动相:以20mmol/L甲酸铵溶液(用甲酸调节pH值至3.6)为流动相A,乙腈(含1%的叔丁基甲醚)为流动相B;按表1进行线性梯度洗脱;流速为1.5mL/min;柱温为35℃;进样体积20μL。西罗莫司主峰的保留时间在23~25min之间。
取西罗莫司对照品约5mg,置于5mL量瓶中,加2mL乙腈使溶解,加0.1mmol/L氢氧化钠溶液1mL,振摇,使混合均匀,加0.1mmol/L盐酸溶液1mL,用乙腈稀释至刻度,摇匀,作为系统适用性溶液。

表1 流动相梯度洗脱程序Tab. 1 Gradient elution program of mobile phase
2.2 专属性试验
经辅料干扰试验,酸破坏,碱破坏,氧化破坏,加热破坏和光照破坏试验知,在建立的色谱条件下,各降解物峰及辅料峰均不干扰断雷帕霉素和西罗莫司的测定,方法专属性良好。典型色谱图见图2。
2.3 线性关系及相对响应因子的测定
分别取西罗莫司对照品和断雷帕霉素对照品各约10mg,精密称定,置于同一100mL量瓶中,加乙腈溶解并稀释至刻度,摇匀,作为混合对照品贮备液。分别精密量取上述储备溶液各适量,用乙腈按不同比例稀释制成浓度分别为0.5、1、2、4、5、10、25和50μg/mL的线性溶液。分别精密量取上述溶液各20μL,分别考察在不同色谱系统及不同色谱柱上的响应情况,记录色谱图。以浓度(C)对峰面积(A)进行线性回归分析。以断雷帕霉素和西罗莫司线性方程的斜率计算断雷帕霉素相对于西罗莫司的响应因子f,其中f为断雷帕霉素线性方程的斜率值/西罗莫司线性方程的斜率值,详见表2。
上述结果表明,断雷帕霉素和西罗莫司在0.5~50μg/mL浓度范围内线性关系均良好,断雷帕霉素相对于西罗莫司的响应因子在0.9~1.1之间。
由于断雷帕霉素对照品难以大量制备且价格昂贵。因此,本文采用西罗莫司加碱破坏的色谱图作为标准图谱,对断雷帕霉素进行定位,并采用主成分自身对照法定量。
2.4 溶液的制备
2.4.1 供试品溶液
取本品适量,精密称定,加乙腈稀释制成约含西罗莫司0.5mg/mL的溶液,滤过,取续滤液作为供试品溶液。
2.4.2 对照溶液
取西罗莫司对照品适量,精密称定,加乙腈溶解并定量稀释制成每5μg/mL的溶液,作为对照溶液。
2.5 溶液稳定性试验
取A企业样品,按“2.4.1”项下制备供试品溶液,在4℃分别放置0、2、4、8、12和24h后,按“2.1”项下的色谱条件进行测定,计算断雷帕霉素的含量,6次测定结果的RSD为1.1%。结果表明,供试品溶液在4℃放置24h内基本稳定。
2.6 重复性试验
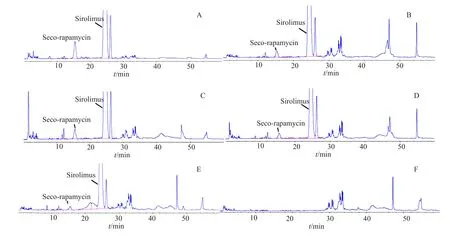
图2 专属性试验HPLC色谱图Fig.2 HPLC Chromatograms of specific test

表2 断雷帕霉素相对响应因子的测定结果Tab. 2 Determination results of the relative response factor
取A企业样品,共6份,按“2.4.1”项下制备供试品溶液,分别进样分析,断雷帕霉素量的平均值为1.0%,RSD为1.4%。重复性良好。
2.7 精密度试验
取“2.3”项下0.5μg/mL的对照溶液,精密量取20μL,分别连续注入液相色谱仪6次,记录色谱图。西罗莫司峰面积的RSD为0.6%。精密度良好。
2.8 检测限
精密量取“2.3”项下线性对照品储备溶液适量,加乙腈逐级稀释,进样分析。以信噪比S/N=3:1计算检测限。检测限为2.3ng(以进样20μL计),约相当于0.02%。
2.9 样品测定
按“2.1”项下的色谱条件,精密量取“2.1”项下的系统适用性试验溶液20μL,注入液相色谱仪,西罗莫司主峰的保留时间约为24min,相对西罗莫司主峰保留时间约为0.6处的色谱峰为断雷帕霉素峰,其他色谱峰均不干扰西罗莫司及断雷帕霉素的测定。A、B和C企业断雷帕霉素检出量分别为1.0%、1.3%和0.7%,典型色谱图见图3。
3 讨论
3.1 现行注册标准有关物质测定方法的缺陷
西罗莫司口服溶液现行注册标准分别采用液相色谱法和质谱法控制有关物质的量。其中,液相色谱法控制西罗莫司发酵工艺杂质去甲基西罗莫司,不控制开环等降解产物,标准存在缺陷,并且存在严重的辅料干扰现象,测定结果重复性差;质谱法控制本品中的同系物、异构体和降解产物。试验发现:该方法的缺陷有:①开环降解产物为有机酸,流动相体系为乙腈-水,开环降解杂质保留较弱,易受辅料基质效应影响且离子化效率和其他杂质存在差异。②样品基质复杂,测定结果重复性差,多次进样后离子源污染较严重。③质谱方法成本高,通用性不强,方法难以推广。
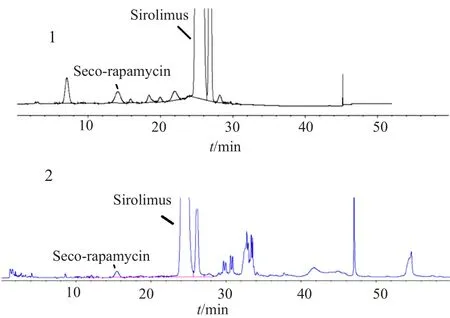
图3 系统适用性试验(1)及供试品溶液(2)典型色谱图Fig.3 HPLCChromatograms of system suitability test (1) and sample solution (2)
3.2 色谱系统建立的依据
由于西罗莫司及其制剂在各国药典中均未收载,在比较了现行注册标准及文献[6-8]调研的基础上,通过溶解样品溶剂的选择、柱温、流动相系统的优化(包括流动相组成、流动相pH、缓冲溶液的选择、流动相中改性剂、互补分离模式的验证等)等试验过程,最终新建专属性良好的,可同时控制西罗莫司开环降解杂质和主要工艺杂质的液相色谱系统[5],为西罗莫司口服溶液色谱系统的建立奠定了基础。
3.3 排除辅料对有关物质测定影响的尝试
西罗莫司口服溶液为黄色澄清黏稠液体。主要辅料为Phosal 50PG,其为复配磷脂,主要为含有溶于丙二醇/乙醇中的约50%磷脂的脂质体形成的组合物[9-10]。由于Phosal组合物具有类似蜂蜜一样的黏稠度。在分析过程中不可避免会产生辅料干扰问题。
试验发现:采用西罗莫司原料有关物质检查方法[5]时,需要在每次进样后用乙腈冲洗色谱系统30min以上,并且辅料的色谱保留“贯穿”整个色谱分析过程。为优化分析方法,尝试沉淀离心去除磷脂的方法,但辅料仍干扰部分工艺杂质及开环降解杂质的测定;尝试采用正相色谱分析系统,利用辅料在正相色谱系统中难保留的特点排除干扰。但是,多次进样后,柱效显著下降,需要活化色谱柱后才能继续检测;通过正相色谱分析系统建立的思路,尝试通过固相萃取的方式,除去辅料的干扰,但部分辅料仍干扰先于西罗莫司色谱峰洗脱的杂质的测定。故上述方法均不是口服溶液有关物质检测的理想方法。
3.4 以断雷帕霉素的量表征西罗莫司口服溶液质量的合理性分析
在西罗莫司分析方法建立的过程中,试验发现:西罗莫司在酸、碱、加热等条件下均不稳定,在相对保留时间约为0.6处均存在较明显的降解色谱峰,经质谱分析及对照品验证,确认该色谱峰为断雷帕霉素。
经稳定性试验研究,断雷帕霉素是主要的开环降解杂质,并且其检出量的增加趋势和西罗莫司含量的降低呈相关性,详见表3~4。因此,拟采用通过控制口服溶液中降解杂质—断雷帕霉素量的方式间接控制西罗莫司口服溶液的质量。为了规避辅料的干扰,在西罗莫司原料有关物质测定方法的基础上对流动相pH值进行调整,并且增加了梯度过程中乙腈的洗脱时间。试验证明,新建的方法操作方便,重复性好,通用性强。
3.5 断雷帕霉素色谱峰定位及定量方式的选择
由于断雷帕霉素对照品难以大量制备,且市场上购置的断雷帕霉素价格昂贵。在没有对照品的前提下,为了更好地实现对断雷帕霉素的定位及后续的定量分析,根据西罗莫司原料方法专属性研究中西罗莫司在碱性条件下易降解产生断雷帕霉素的现象,在方法中增加碱破坏试验作为系统适用性溶液,并附标准图谱,明确断雷帕霉素的保留行为。由于断雷帕霉素相对于西罗莫司的响应因子在0.9~1.1之间,因此以主成分自身对照法定量,提高定量结果的准确性。
4 小结
现行西罗莫司口服溶液标准均存在一定的缺陷,难以真实反映产品的质量。本文通过色谱系统的筛选及流动相条件的优化,自建了以主要开环降解杂质断雷帕霉素的量表征西罗莫司口服溶液质量的方法,为完善质量标准,有效监控产品的质量奠定了基础。
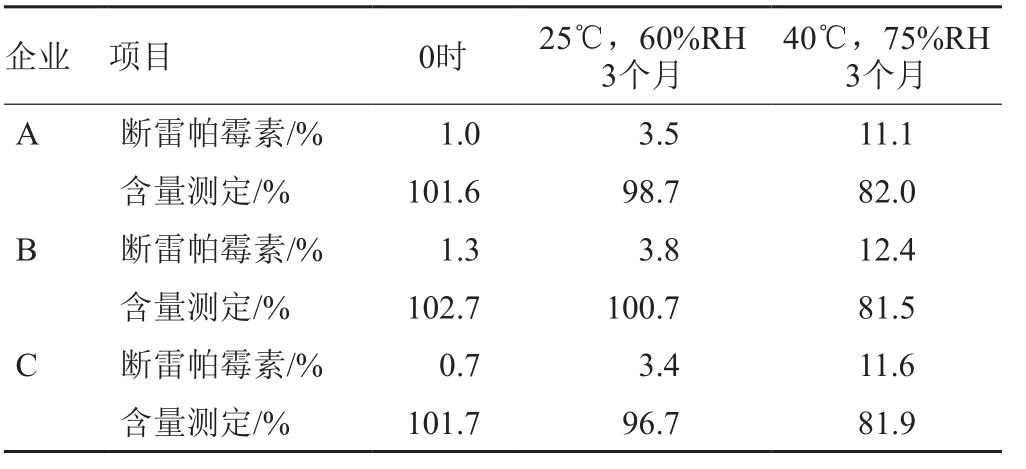
表3 稳定性试验断雷帕霉素量和西罗莫司含量结果Tab. 3 Stability test results of seco-rapamycin and the content of sirolimus

表4 断雷帕霉素和西罗莫司测定结果相关分析Tab. 4 Correlation analysis result between seco-rapamycin and the content of sirolimus
将开环降解杂质断雷帕霉素作为表征制剂工艺及贮藏过程中影响产品质量的特定杂质,解决了西罗莫司口服溶液中辅料对检测造成的严重干扰问题。此方法专属性、重复性好,简便易行,为复杂体系样品中有关物质的考察提供了新的思路和方法。
