分散固相萃取-超高效液相色谱-串联质谱法测定奶粉中7种非选择性环氧化酶抑制药物
2019-01-29金朦娜王红青邱红钰
励 炯, 孙 岚, 金朦娜, 王红青, 邱红钰
(杭州市食品药品检验研究院, 浙江 杭州 310017)
非甾体类抗炎药物(nonsteroidal anti-inflammatory drugs, NSAIDs)是一大类具有抗炎、止痛和解热等疗效的药物总称,其中有一类属于非选择性环氧化酶(cyclooxygenase, COX)抑制药物,其作用机理是通过抑制COX的活性来抑制前列腺素的分泌,从而缓解肌肉和关节的局部疼痛、肿胀等症状[1,2]。非选择性COX抑制药物已被广泛用于人和牲畜的疾病预防与治疗[3],但对人体和牲畜也有诸多毒副作用,能引起肠胃道功能紊乱、过敏、再生障碍性贫血、凝血功能障碍、肝肾毒性、心血管疾病以及中枢神经系统损害等[4-6]。有研究[7]表明,长期使用非选择性COX抑制药物会引起大鼠的肾脏癌变和小鼠的肝脏癌变。欧盟、美国和日本[8]已针对部分非选择性COX抑制药物在动物源性食品中的最大残留限量做了规定。
非选择性COX抑制药物是由不同化学结构组成的一类化合物,很多化合物化学性质完全不同,这就带来一个同时检测多种非选择性COX抑制药物的方法开发的难题,特别是样品前处理(包括提取和净化)[9]。目前,已报道的非选择性COX抑制药物的检测方法主要有高效液相色谱法[10,11]、液相色谱-质谱联用法[12-15]、气相色谱-质谱联用法[16,17]、毛细管电泳法[18]、生物传感器法[19]等。液相色谱-质谱联用法由于具有较高的灵敏度和选择性,非常适合多种药物残留的检测,已经被广泛应用于非选择性COX抑制药物的检测[20,21],比如Gallo等[22]报道了利用串联质谱法同时检测人体血清中16种非选择性COX抑制剂的方法;Dubreil-Chéneau等[13]和Gentili等[18]利用串联质谱法分别开发了牛乳中12种和15种非选择性COX抑制药物残留的检测方法;Dowling等[23]利用高分辨质谱法检测牛乳中8种非选择性COX抑制药物残留。上述方法的前处理多用甲醇或乙腈进行提取,再经液液萃取净化或SPE固相萃取小柱净化处理,缺点是在大批量检测中耗时较长。本文采用分散固相萃取(dSPE)对奶粉中非选择性COX抑制药物进行净化,并利用灵敏度高和抗干扰性强的超高效液相色谱-串联质谱法,对奶粉中7种非选择性COX抑制药物进行定性定量分析。
1 实验部分
1.1 仪器与试剂
超高效液相色谱-串联四极杆质谱联用仪(包括Agilent 1290高效液相色谱仪和Agilent 6425串联四极杆质谱仪,美国Agilent公司)、Milli-Q去离子水发生器(美国Millipore公司)、Sorvall ST 8R高速冷冻离心机(德国Thermo Fisher公司)、MS3旋涡混合器(德国IKA公司)。
标准品:水杨酸(批号100106-201605,质量分数99.7%)、布洛芬(批号100179-201406,质量分数99.5%)、双氯芬酸钠(批号100334-200302,质量分数100%)、吲哚美辛(批号100528-200904,质量分数99.9%)、吡罗昔康(批号100177-200603,质量分数99.8%)、萘普生(批号100198-201205,质量分数99.6%)、保泰松(批号100481- 200601,质量分数99.9%)购自中国食品药品检定研究院;盐酸、抗坏血酸、氯化钠、无水硫酸镁(均为分析纯)购自国药集团化学试剂有限公司;乙腈、甲酸、乙酸乙酯(均为色谱纯)购自德国Merck公司;十八烷基键合硅胶吸附剂(C18-N)和NH2-丙基乙二胺吸附剂(NH2-PSA)购自上海岛津技迩公司。
1.2 标准溶液的配制
分别取7种非选择性COX抑制药物的标准品10 mg,用乙腈配制成1 000 mg/L的标准储备溶液,于-20 ℃冰箱中保存,备用。
分别吸取适当体积的标准品储备溶液,用乙腈稀释成10 mg/L的混合标准中间溶液,于-4 ℃冰箱中保存,备用。
用乙腈稀释混合标准中间溶液,配制成水杨酸、吲哚美辛、吡罗昔康和保泰松的线性范围为5~200 μg/L,萘普生、双氯芬酸钠和布洛芬的线性范围为10~200 μg/L的溶剂混合标准工作溶液,现用现配。
1.3 质控样品的配制
取2.0 g空白奶粉(检测结果为阴性的奶粉),置于50 mL聚氟乙烯离心管中,加入一定量的混合标准中间溶液,使7种非选择性COX抑制药物在样品中的质量浓度为50 μg/kg,即为质控样品。
1.4 样品前处理
提取:取2.0 g奶粉,置于50 mL聚氟乙烯离心管中,加入5 mL去离子水,涡旋振荡2 min,使样品充分溶解,加入2 mL 0.01 mol/L的抗坏血酸溶液(用1 mol/L盐酸溶液调pH至2.5),涡旋振荡30 s,依次加入10 mL乙腈-乙酸乙酯(1∶1, v/v)溶液、1 g醋酸铵和5 g无水硫酸镁,涡旋振荡1 min,超声提取5 min,在4 ℃以5 000 r/min的速度离心5 min。取7 mL上清液移入新的50 mL聚氟乙烯离心管中,待净化。
净化:在待净化的样品溶液中加入净化剂(含1 000 mg无水硫酸钠、300 mg C18-N及100 mg NH2-PSA),旋涡混匀1 min, 以5 000 r/min的速度离心5 min。精密取5 mL上清液转移至试管中,于40 ℃水浴下氮吹至近干。加入0.5 mL 0.1%(体积分数,下同)甲酸水溶液-乙腈(1∶1, v/v),旋涡混匀1 min,溶解残渣,用0.22 μm有机滤膜过滤,取滤液进行分析。
1.5 色谱和质谱条件
色谱柱:Waters CORTECS C18(100 mm×2.1 mm, 1.7 μm);流动相:A相为0.1%甲酸水溶液,B相为0.1%甲酸乙腈。梯度洗脱程序:0~8.0 min, 90%A~45%A; 8.0~9.0 min, 45%A; 9.0~9.1 min, 45%A~90%A; 9.1~12.0 min, 90%A。柱温:40 ℃;流速:0.4 mL/min,进样量:1 μL。
离子源:电喷雾离子(ESI+和ESI-)源;多反应监测(MRM)模式;干燥气温度:200 ℃;干燥气流速:14 L/min;毛细管电压:3.5 kV;毛细管出口电压(fragmentor): 380 V;校准方法:质量轴自动调谐校正。MRM进行分段扫描:0~4.50 min,水杨酸;4.50~6.00 min,吡罗昔康;6.00~7.50 min,萘普生;7.50~8.85 min,吲哚美辛、双氯芬酸钠和布洛芬;8.85~12.00 min,保泰松。其他质谱参数详见表1。
2 结果与讨论
2.1 质谱和色谱条件的优化
将7种非选择性COX抑制药物的标准品溶液(1 mg/L)直接进样,分别在ESI+和ESI-两种模式下进行一级质谱扫描,结果表明,水杨酸、萘普生、吲哚美辛、双氯芬酸钠、布洛芬在ESI-模式下的响应比较高,而吡罗昔康和保泰松两种成分在ESI+模式下的灵敏度较高。得到7种化合物精确的母离子质荷比,优化毛细管电压。进一步对7种非选择性COX抑制药物的二级质谱参数进行优化,选择信号强度较大的2个碎片离子为特征子离子,其中相对丰度最强的为定量离子,其次为定性离子,以MRM模式进行扫描,详细参数见表1。其中布洛芬的碎片离子只有一个,所以采用一个离子通道对奶粉中的布洛芬进行分析。

表 1 7种非选择性COX抑制药物的质谱参数
* Quantitative ion pair.
考察了7种非选择性COX抑制药物在3组不同流动相下的色谱行为:(1)A为0.1%甲酸水溶液,B为0.1%甲酸甲醇;(2)A为0.1%甲酸水溶液,B为0.1%甲酸乙腈;(3)A为0.1%甲酸水溶液(含5 mmol/L甲酸铵), B为0.1%甲酸乙腈。研究发现,在流动相(1)条件下,7种非选择性COX抑制药物的分析时间过长,超过30 min,而且色谱峰峰形较差;在流动相(2)条件下,7种非选择性COX抑制药物的分析时间短,各组分色谱峰峰形较好,灵敏度满足要求。本文考虑到7种非选择性COX抑制药物中除了吡罗昔康和保泰松,其余5种均为是ESI-模式检测,因此在0.1%甲酸水溶液中加入5 mmol/L的甲酸铵(即流动相(3)),来提高负离子模式检测的5种非选择性COX抑制药物的离子化程度,结果发现效果不明显,而且会造成吡罗昔康和保泰松的色谱峰严重拖尾的现象,所以将0.1%甲酸水溶液和0.1%甲酸乙腈作为分析7种非选择性COX抑制药物的流动相体系。
复溶溶剂的比例也会影响残渣中非选择性COX抑制药物的色谱行为和回收率,本文采用0.1%甲酸水溶液-乙腈作为复溶溶剂。对复溶溶剂中乙腈的体积分数(分别为10%、50%和90%)进行了考察,结果表明,当乙腈体积分数为10%时,残渣中保泰松的溶解度较小,回收率较低;当乙腈体积分数为90%时,7种非选择性COX抑制药物的色谱峰会出现前倾和分叉峰;当乙腈体积分数为50%时,7种非选择性COX抑制药物的检测灵敏度最高,色谱峰峰形较好,因此本文采用0.1%甲酸水-乙腈(1∶1, v/v)作为残渣中7种非选择性COX抑制药物的复溶溶剂。7种非选择性COX抑制药物的MRM谱图见图1。
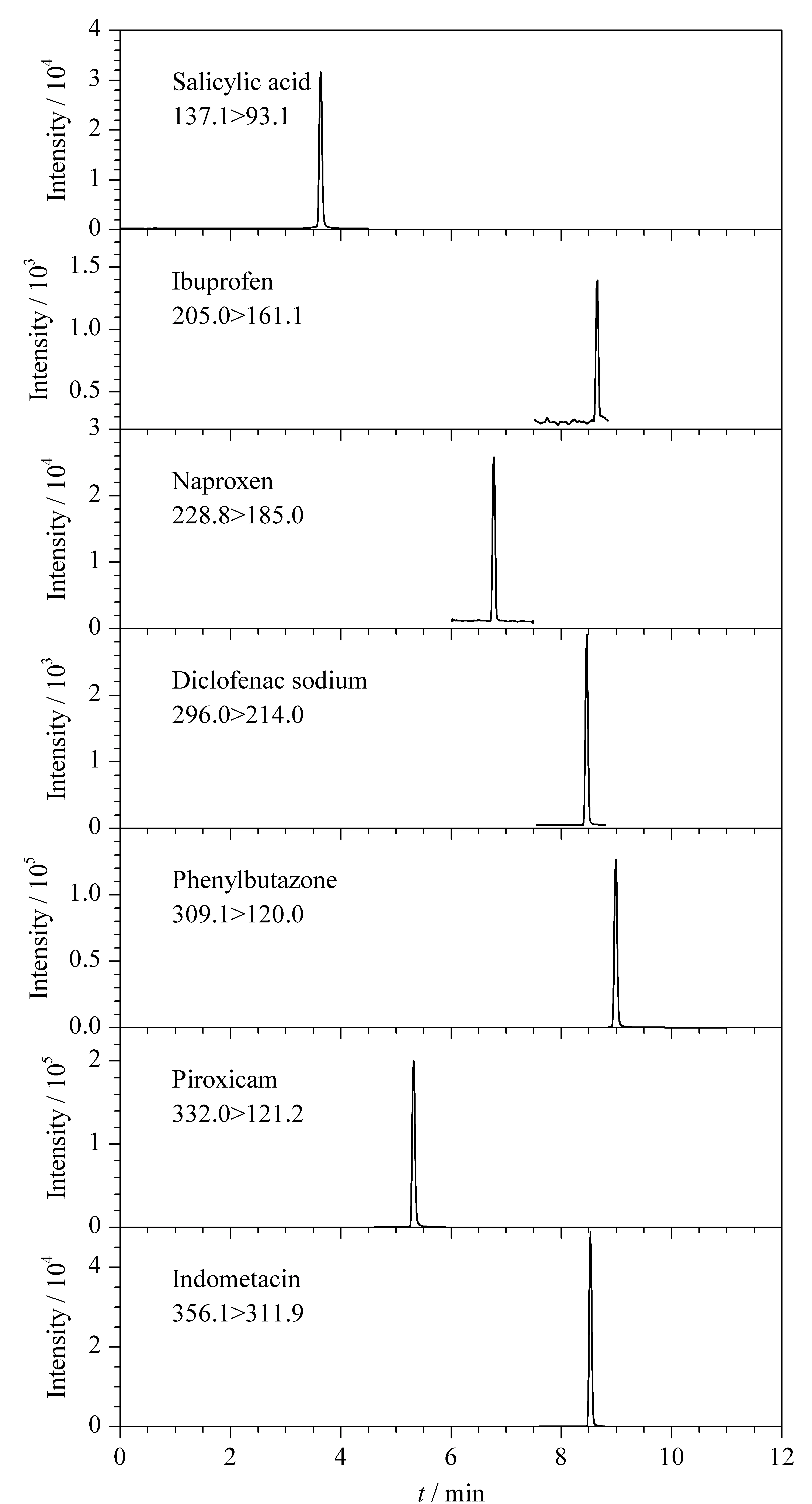
图 1 7种非选择性COX抑制药物(20 μg/L)的MRM谱图Fig. 1 MRM chromatograms of the seven non-selective COX inhibitors (20 μg/L)
2.2 提取方式的优化
非选择性COX抑制药物常用的提取剂是甲醇和乙腈,除了其高效的提取效率之外,还具有净化效果,可以去除样品中的部分蛋白质。有文献[21]报道用5%甲酸乙腈(酸化乙腈)提取牛乳中的非选择性COX抑制药物,提取效率较高,但是由于酸化乙腈仅仅对极性较强的化合物提取效果好,对于弱极性或者非极性化合物的提取效率一般。有文献[8]采用乙腈-乙酸乙酯(1∶1, v/v)溶液提取牛乳中的非选择性COX抑制药物,因为乙酸乙酯能够提高弱极性或者非极性化合物(包括保泰松、萘普生、吡罗昔康、布洛芬和吲哚美辛)提取效率。所以本文将乙腈-乙酸乙酯(1∶1, v/v)溶液作为奶粉中非选择性COX抑制药物的提取溶剂。有研究[16]发现保泰松在前处理过程中容易被氧化,回收率和重现性不高,需要在提取过程中加入一定量的抗坏血酸来防止保泰松的氧化。也有研究[21]表明,在提取过程中加入高浓度(0.1 mol/L)的抗坏血酸,会导致萘普生回收率降低,且并不能显著提高保泰松的回收率,同时会污染质谱系统。所以本文将抗坏血酸的浓度降至0.01 mol/L,并用1 mol/L盐酸溶液将pH调至2.5,结果表明,采用0.01 mol/L pH 2.5的抗坏血酸溶液-乙酸乙酯-乙腈(2∶5∶5, v/v/v)组成的提取溶剂提取质控样品中7种非选择性COX抑制药物,保泰松的提取效率最高,而且不影响其他6种化合物的提取效率。提取溶剂的酸化处理(加入pH 2.5的抗坏血酸)能充分去除提取溶液中的蛋白质,还能增加非选择性COX抑制药物的pKa值,促进其质子化作用[8]。不同提取溶剂对奶粉中7种非选择性COX抑制药物的提取效率见图2。
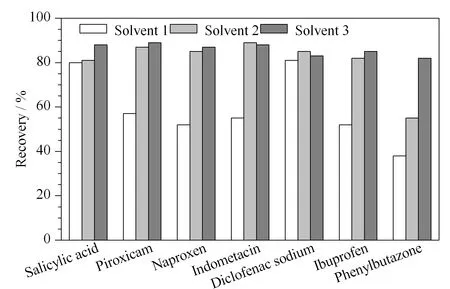
图 2 不同提取溶剂对质控样品中7种非选择性COX 抑制药物的提取效率Fig. 2 Extraction recoveries using different extraction solutions in quality-control samples Solvent 1∶ 5% (volume percentage) formic acid acetonitrile solution; solvent 2: acetonitrile-ethyl acetate (1∶1, v/v); solvent 3∶ 0.01 mol/L pH 2.5 ascorbic acid-acetonitrile-ethyl acetate (2∶5∶5, v/v/v).
2.3 净化方式的优化
有报道[13]将牛乳中的非选择性COX抑制药物经甲醇提取后直接进样,但是考虑到奶粉中高含量的脂类物质,会引起基质效应,影响检测灵敏度,污染色谱和质谱系统,所以有必要对提取溶液进行净化处理。检测奶制品中的非选择性COX抑制药物的净化步骤一般采用两种方式:液液萃取净化和固相萃取小柱净化(SPE)[18,23]。Peng等[8]用正己烷去除提取液中的脂类物质。而最常用的净化方式是用SPE固相萃取小柱,已有的文献[22]采用C18、亲水亲脂共平衡的大孔聚合物(HLB)、混合型阳离子交换树脂(MCX)等填料的固相萃取小柱去除样品中的脂类物质,但Dubreil-Chéneau等[13]用固相萃取小柱净化后,发现结果重现性很差。
本文采用分散固相萃取对提取溶液进行净化,采用无水硫酸钠、C18-N及NH2-PSA 3种净化剂,对其用量进行正交试验优化。无水硫酸钠、C18-N及NH2-PSA用量分别作为因素A、B、C,每个因素取3个水平,按照L9(33)正交表试验,比较质控样品在各因素组合下的回收率和净化效果,来选择最佳配比组合(见表2)。结果表明,使用1 000 mg无水硫酸钠、300 mg C18-N以及100 mg NH2-PSA组成的净化剂可以获得良好的净化效果。

表 2 无水硫酸钠、C18-N及NH2-PSA用量的正交试验因素水平

表 3 7种非选择性COX抑制药物的线性范围、线性方程、r2、检出限和定量限
Y: peak area of quantitative ion;X: mass concentration, μg/L.
本文通过比较分散固相萃取、正己烷液液萃取和固相萃取(采用C18、HLB、MCX等3种填料)等3种净化方式,发现质控样品的净化效率差别不大。考虑到实验的成本、效率和结果的重现性,最终选择分散固相萃取作为检测奶粉中7种非选择性COX抑制药物的净化方式。
2.4 基质效应考察
样品前处理过程中,除了目标化合物,还有其他同时提取出来的干扰成分,这些共流出组分会影响目标化合物的分离并干扰其离子化,引起检测信号的减弱或增强,最终影响定量的准确性,这就是基质效应(ME)。一般用基质标准曲线的斜率与溶剂标准曲线的斜率之比作为ME,评价前处理的净化效率。一般情况下,ME在85%~115%之间不存在明显的基质效应。本文采用空白奶粉的提取溶液作为基质,分别精密加入适当体积的混合标准中间溶液,配制成水杨酸、吲哚美辛、吡罗昔康和保泰松的线性范围为5~200 μg/L以及萘普生、双氯芬酸钠和布洛芬的线性范围为10~200 μg/L的基质匹配标准工作液;溶剂混合标准工作溶液配制见1.2节。按1.5节仪器条件进行分析,得到基质标准曲线和溶剂标准曲线,结果ME为77.6%~87.1%,表明存在基质抑制效应,为消除或减弱基质效应干扰,本文采用相应空白基质溶液配制标准溶液绘制校正曲线。
2.5 线性关系、定量限及重复性
取2.4节中已配制的基质匹配标准工作液,分别进样1 μL,以定量离子峰面积(Y)为纵坐标、标准品质量浓度(X, μg/L)为横坐标进行线性回归,得到线性方程、线性范围和相关系数。结果表明,各组分在线性范围内线性关系良好,相关系数(r2)均大于0.995 7。
取2.0 g空白样品(检测结果为阴性的奶粉),加入适当稀释的混合标准中间溶液,按1.4节方法进行前处理,按1.5节方法进行分析。如表3所示,以定量离子对的信噪比(S/N)=3为检出限(LOD)、S/N=10为定量限(LOQ),得到检出限范围为0.5~2 μg/g,定量限范围为2~5 μg/g。
2.6 方法回收率与精密度
取2.0 g空白样品(检测结果为阴性的奶粉),添加低、中、高3个水平(详见表4),每个水平有6份样品,回收率和精密度试验结果见表4。7种非选择性COX抑制药物的3个浓度水平的平均回收率为76.4%~89.8%,相对偏差为2.1%~7.9%。

表 4 奶粉中7种非选择性COX抑制药物的回收率与精密度试验(n=6)
2.7 实际样品检测
用本方法完成了40批次奶粉(均来自2017年度杭州地区奶粉专项抽检)的检测,结果均未检出7种非选择性COX抑制药物。
3 结语
本文建立了分散固相萃取-超高效液相色谱-串联质谱测定奶粉中7种非选择性COX抑制药物残留的方法,优化了色谱、质谱、提取和净化条件。与传统固相萃取技术相比,该方法更加简便、经济、高效,能够满足日常大批量奶粉检测的需求。
