固相萃取-大体积程序升温进样气相色谱-三重四极杆串联质谱测定饮用水中3种挥发性N-亚硝胺
2019-01-29李登昆刘祥萍赵士权陈春静熊丽林
李登昆, 张 云, 刘祥萍, 赵士权, 陈春静, 熊丽林
(南京市疾病预防控制中心, 江苏 南京 210003)
N-亚硝胺来源于橡胶、皮革等化工制造业,同时也是饮用水消毒副产物(disinfection by-product, DBP),对哺乳动物具有强烈的遗传毒性及致癌性[1-4]。国际癌症研究机构(international agency for research on cancer, IARC)将N-亚硝基二甲基胺(N-nitrosodimethylamine, NDMA)、N-亚硝基甲基乙基胺(N-nitrosomethylethylamine, NMEA)列为2A类致癌物,将N-亚硝基二乙基胺(N-nitrosodiethylamine, NDEA)列为2B类致癌物[5]。世界卫生组织建议饮用水中NDMA限量为100 ng/L[6],美国环境保护署(USEPA)规定饮水中NDMA、NMEA、NDEA最大容许浓度分为7、20及2 ng/L[7]。
自1989年加拿大安大略省[8]确认饮用水中消毒副产物NDMA以来,各国日益重视饮用水中N-亚硝胺问题,而建立灵敏、准确、高效、简便的检测方法也成为研究者关注的重点。目前国内外针对挥发性N-亚硝胺的检测主要采用气相色谱(GC)法,常用检测器有氢火焰离子化检测器(FID)、氮磷检测器(NPD)、热能分析(TEA)仪及单四极杆质谱(Q-MS)。FID[9,10]是通用型检测器,定性能力差、方法灵敏度低,NPD[9,11]对N-亚硝胺的检测也存在响应低的缺点,随着新技术的发展FID、NPD逐渐被取代;TEA[9,12]对N-亚硝胺具有高度特异性,但应用范围窄且价格较为昂贵;Q-MS[9,12,13]是早期检测N-亚硝胺的主要方式,性能稳定、应用广泛,但对EI源产生无差别的离子碎片甄别困难、假阳性干扰难以排除。近年来随着三重四极杆质谱联用仪(QqQ-MS/MS)的发展,气相色谱与三重四极杆质谱仪联用已逐渐成为N-亚硝胺分析的常规技术手段,其采用多反应监测(MRM)模式扫描,具有选择性好、定性特异性强、定量准确度高的优点[9,14-18]。

表 1 3种挥发性N-亚硝胺的理化性质
NDMA:N-nitrosodimethylamine; NMEA:N-nitrosomethylethylamine; NDEA:N-nitrosodiethylamine; IARC: International Agency for Research on Cancer; 1 mmHg=0.133 kPa; logKow: octanol-water partition coefficient; logKoc: organic carbon partition coefficient.
由于饮用水中N-亚硝胺的含量极低,多在ng/L的水平[4,19],如何提取、浓缩是困扰研究者的另一难题,目前国内外基本都采用固相萃取再氮吹的方式对饮用水中亚硝胺进行富集、浓缩[9,14,20,21,24],但NDMA、NMEA及NDEA的沸点低、蒸汽压大、挥发性强,氮吹浓缩极易损失,即便采取添加同位素内标的方式,回收率依然难以达到满意效果[14,20,21]。大体积程序升温进样(LVI-PTV)技术使用容纳大体积样品的装置及可控时间的溶剂放空装置,选择性去除样品中的大量溶剂,可以实现痕量目标化合物的在线浓缩,避免挥发性物质的损失,简化操作步骤,缩短样品预处理时间,同时提高分析方法的灵敏度、精密度及准确度[22,23]。固相萃取-程序升温进样技术结合气相色谱-三重四极杆质谱联用仪,将是分析饮用水中易挥发痕量组分的理想手段。3种挥发性N-亚硝胺的理化性质见表1。
1 实验部分
1.1 仪器与设备
气相色谱-三重四极杆质谱联用仪(Agilent 7890B/7000C,美国Agilent公司),配备Agilent PAL自动进样系统及Agilent多模式进样口(MMI);全自动固相萃取仪(Thermo Fisher AT280,美国Thermo Fisher公司); Milli-Q水纯化系统(Milli-Q Reference,美国Millipore公司);电子分析天平(ML203/02,瑞士METTLER TOLEDO公司)。
1.2 试剂与材料
N-亚硝胺混合标准品包括NDMA(CAS: 62-75-9)、NMEA(CAS: 10595-95-6)和NDEA(CAS: 55-18-5),纯度均大于99.5%,质量浓度均为100 mg/L,购自美国o2si公司;甲醇(色谱纯)购自德国Merck公司;二氯甲烷(色谱纯)和椰壳活性炭SPE小柱(2 g/6 mL, 80~120目)购自德国CNW公司;无水硫酸钠(分析纯,使用前于马弗炉中450 ℃灼烧4 h,冷却后密封保存备用)和硫代硫酸钠(分析纯)购于国药集团化学试剂限公司;纯水由Milli-Q净化系统制得。
1.3 仪器条件
1.3.1色谱条件
大体积程序升温进样口,进样方式:溶剂排空进样;进样体积:10 μL;隔垫吹扫流量:3 mL/min;升温程序:50 ℃保持0.08 min,以600 ℃/min升至325 ℃,保持5 min;溶剂放空模式:到分流出口的吹扫流量:60 mL/min;吹扫时间:2.58 min;放空流量:100 mL/min;放空时间:0.08 min;载气节省模式下氦气流速:20 mL/min,等待时间:5 min。
色谱柱:VF-WAXms柱(30 m×0.25 mm×0.25 μm,美国Agilent公司);载气:He(纯度>99.999%);流量:1.2 mL/min。
柱温升温程序:初始温度为50 ℃,保持1 min,以10 ℃/min的速度升温至110 ℃,再以15 ℃/min的速度升温至200 ℃,最后以40 ℃/min的速度升温至250 ℃,保持7 min。
1.3.2质谱条件
离子源:EI源;传输线温度:250 ℃;离子源温度:280 ℃;电离能量:70 eV;前四极杆温度:180 ℃;后四极杆温度:180 ℃;碰撞池:碰撞气为N2(纯度>99.999%),流速为1.5 mL/min;淬灭气为He(纯度>99.999%),流速为2.25 mL/min;扫描方式:MRM模式;溶剂延迟:5 min。3种挥发性N-亚硝胺质谱参数见表2。
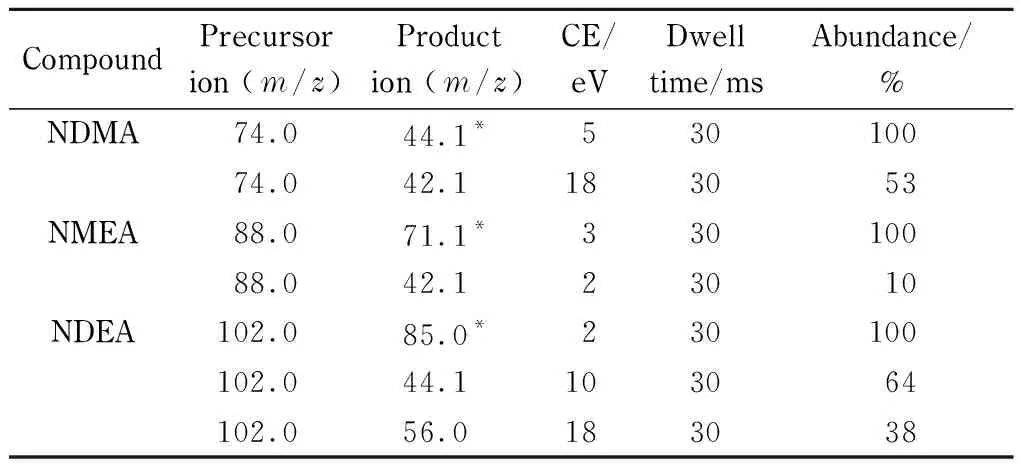
表 2 3种挥发性N-亚硝胺的前级离子、子离子、碰撞能量(CE)、驻留时间及丰度
* Quantitative ion.
1.4 标准溶液的配制
将100 mg/L标准溶液从-20 ℃冰箱取出,置室内恒温后,用甲醇稀释,配制成2.0 mg/L的标准使用液,置于棕色玻璃瓶内,再用二氯甲烷逐步稀释,配制成质量浓度为100、200、500、1 000、2 000和5 000 ng/L的系列标准溶液。
1.5 样品处理
椰壳活性炭SPE小柱依次用6 mL二氯甲烷冲洗并用氮气吹干,用6 mL甲醇冲洗并用氮气吹干,用6 mL甲醇及15 mL纯水活化平衡,活化平衡过程连续进行并保证SPE小柱液面不干。将1.0 L水样以10 mL/min的速度通过SPE小柱,萃取完成后氮吹10 min,吹干SPE小柱。用10 mL二氯甲烷分2次(每次5 mL)浸泡洗脱至收集管,洗脱液加入5~8 g无水硫酸钠脱水,取1.0 mL萃取液上机测定。
2 结果与讨论
2.1 色谱柱的选择
对比了DB-5MS Ultra Inert柱(30 m×0.25 mm×0.25 μm,美国Agilent公司)与VF-WAXms(30 m×0.25 mm×0.25 μm)两种色谱柱的分离能力,结果显示,3种挥发性N-亚硝胺在两种色谱柱上于11 min内都能获得良好的分离,但使用VF-WAXms色谱柱可获得更好的峰形和灵敏度,故本法选择VF-WAXms色谱柱作为分析柱。两种色谱柱分离效果见图1。
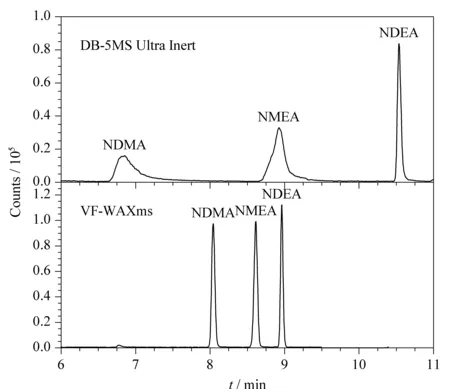
图 1 使用DB-5MS Ultra Inert柱和VF-WAXms 柱时3种挥发性N-亚硝胺(10 μg/L)在MRM模式下的总离子流图 Fig. 1 Total ion chromatograms (TIC) of the three volatile N-nitrosamines (10 μg/L) obtained using the DB-5MS Ultra Inert column and VF-WAXms column in MRM mode DB-5MS Ultra Inert column: initial 35 ℃, hold for 5 min; increase to 250 ℃ at a rate of 20 ℃/min, hold for 5 min; VF-WAXms column: initial 50 ℃, hold for 1 min; increase to 110 ℃ at a rate of 15 ℃/min; rise to 250 ℃ at a rate of 40 ℃/min, hold for 7 min.
2.2 质谱条件的优化
N-亚硝胺组分经质谱仪离子化后,通过母离子、子离子扫描方式,筛选出较理想的多反应监测(MRM)离子对,再结合Agilent MassHunter设计实验助手与分析实验助手软件,优化选择出最佳离子对、碰撞能量及驻留时间等,3种挥发性N-亚硝胺最佳MRM质谱参数见表2, MRM色谱图见图2。

图 2 3种挥发性N-亚硝胺混合标准品(10 μg/L) 的MRM色谱图Fig. 2 MRM chromatograms of the three volatile N-nitrosamines mixed standard (10 μg/L)

CompoundRT/minLinear range/(ng/L)Linear equationr2LOD/(ng/L)LOQ/(ng/L)NDMA8.0451-50Y=22.992X+30.5130.99920.20.7NMEA8.6281-50Y=18.486X+7.0200.99910.060.2NDEA8.9721-50Y=15.437X+9.7730.99990.030.08
Y: peak area;X: mass concentration, ng/L.
2.3 大体积程序升温进样的优化
大体积程序升温气化进样,通过增加进样量来提高方法灵敏度,但增大进样量的同时也会引入更多的“脏”物质进入色谱系统,造成色谱柱及离子源的污染,故在满足方法灵敏度的条件下,应尽量减少进样体积。本文通过多次试验优化进样量,当进样体积为10 μL时,可满足USEPA Method 521[14]最低检出浓度要求,然后考查初始温度、排空时间、升温速率等各变量因素,大体积程序升温进样最佳条件见1.3.1节。
2.4 固相萃取柱的选择
饮用水中痕量N-亚硝胺的萃取、富集,目前最常用的吸附材料为椰壳活性炭[14,20,24],本文采用80~120目椰壳活性炭SPE小柱进行饮用水添加回收试验,取得较好试验预期,结果见表3。有文献[16]报道采用Oasis HLB固相萃取小柱萃取化妆品中10种挥发性亚硝胺取得满意效果,但本文采用该固相萃取小柱对饮用水进行加标回收试验,3种挥发性N-亚硝胺均未能有效萃取,这与朱翔[24]等人的实验结果基本一致。同时,还对C18固相萃取小柱进行了考查,同样无法有效萃取3种N-亚硝胺。
2.5 洗脱溶剂的选择及洗脱体积的优化
目前国内外主要研究方法中都选用二氯甲烷作为N-亚硝胺的萃取、洗脱溶剂[14,15,20,21,24,25],且相关研究表明二氯甲烷是最理想的洗脱溶剂[25],故本文选择二氯甲烷作为洗脱溶剂,并考查了二氯甲烷洗脱体积对洗脱效率的影响。在饮用水加标浓度为50 ng/L时,分别用5、10和15 mL的二氯甲烷进行洗脱,3种挥发性N-亚硝胺平均洗脱效率分别为73.2%~85.8%、103%~109%和92.6%~99.6%,结果显示,洗脱量为5 mL时,洗脱不够充分,洗脱量为10和15 mL时,均可充分洗脱,为避免浪费试剂、污染环境,选用10 mL作为洗脱体积。
2.6 线性范围与检出限
配制线性范围为1~50 ng/L的系列标准溶液,按1.3节仪器条件测定,以定量离子峰面积(Y)对浓度(X, ng/L)绘制线性回归方程,以3倍信噪比(S/N)为检出限,以10倍S/N为定量限。本法饮用水取样量为1.0 L, 3种挥发性N-亚硝胺组分的保留时间、线性范围、线性方程、相关系数、检出限(LOD)及定量限(LOQ)见表3。
2.7 准确度及精密度
以实验室自来水作为饮用水样,加入1.4节的标准使用液,充分搅拌混匀,配制成10.0、20.0、50.0 ng/L的加标样品,每个水平各5份,按照1.5节样品萃取程序进行预处理,按1.3节仪器条件测定,同时取纯水做空白对照。以加标回收率指标考察方法的准确度及精密度,结果见表4。加标回收率为94.8%~109%、相对标准偏差小于10%,说明该方法准确度和精密度较好。
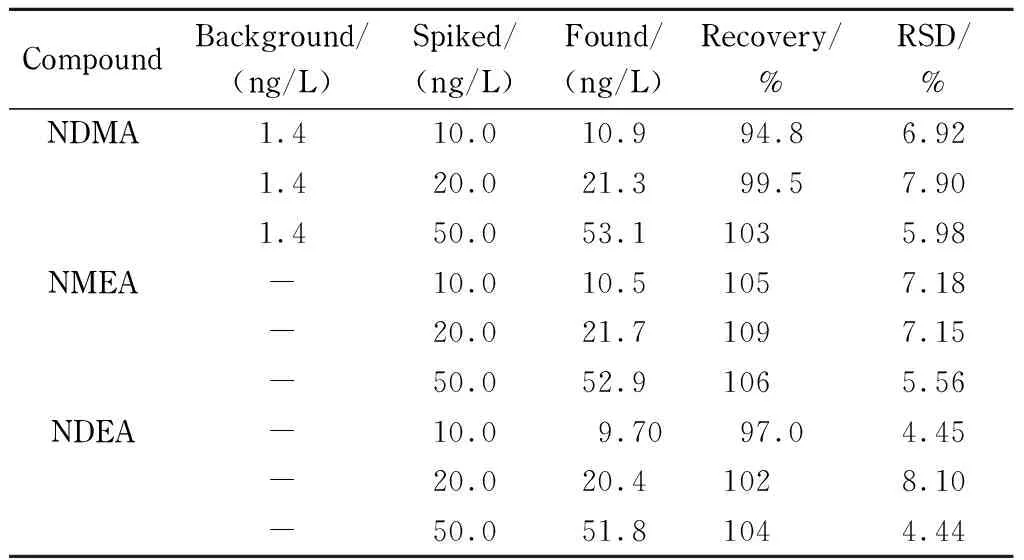
表 4 3种挥发性N-亚硝胺在饮用水中3个添加水平下的加标回收率及精密度(n=5)
-: less than LOQ.
2.8 饮用水样的采集及测定
参照GB/T 5750.2-2006[26]的相关采样规定,选用1 L具聚四氟乙烯材质盖垫棕色螺纹口玻璃瓶采集样品,在采样之前加入0.1 g硫代硫酸钠固体作为脱氯剂,水样充满玻璃瓶后加盖密封,避光、冷藏运输,送达实验室后应及时完成萃取检测。采集本市7家自来水厂的水源水、出厂水、末梢水及市售瓶装饮用天然水、饮用净水共27份,进行饮用水中3种挥发性N-亚硝胺含量的测定。样品测定结果见表5。
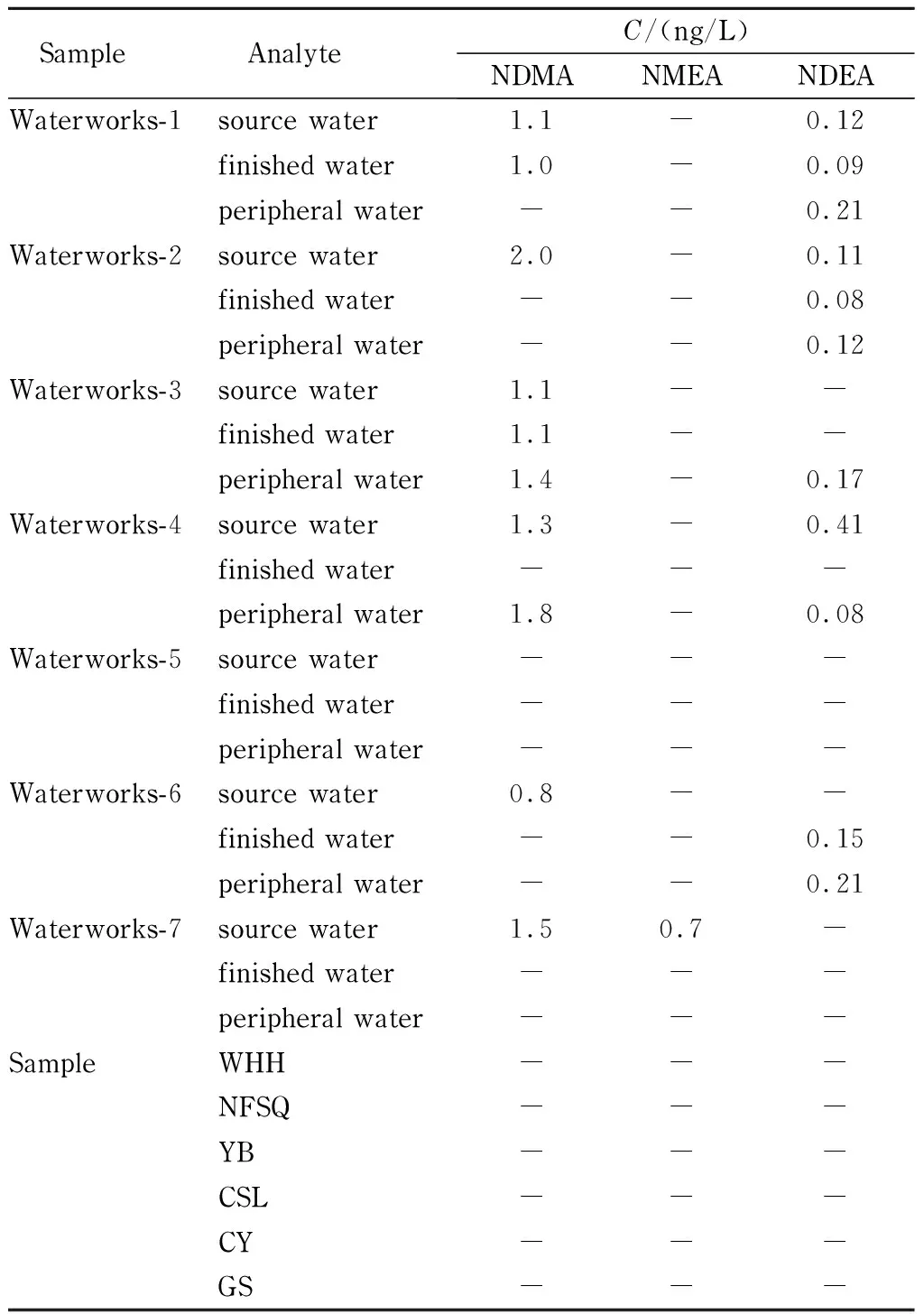
表 5 饮用水中3种挥发性N-亚硝胺含量测定结果
-: less than LOQ. WHH, NFSQ, YB, CSL, CY, and GS stand for acronyms of the brand of bottled drinking water.
3 结论
本文建立了SPE-PTV-GC-EI-MS/MS测定饮用水中3种挥发性亚硝胺的方法,采用保留时间和MRM双重定性、外标法定量,本法选择性好、灵敏度高、样品前处理简单,线性关系、加标回收率及重现性等方法学指标均较好,是饮用水中挥发痕量N-亚硝胺组分理想的检测技术手段。
