动物源性食品中青霉素类药物残留检测
2019-01-08马燕红李志刚李莹莹郭文萍
马燕红,王 妍,李志刚,李莹莹,郭文萍
(中国肉类食品综合研究中心,北京 100068)
近几十年来抗生素,尤其是β-内酰胺类抗生素,由于其在人类医学和兽药学上有效地预防和治疗各种传染性疾病而备受关注。青霉素属于β-内酰胺类抗生素,由于其药效高、价格低廉、使用方便,已成为临床上治疗细菌感染最常用的抗生素,在畜牧兽医水产养殖领域也得到了广泛的应用。目前主要用于治疗奶牛乳腺炎、鱼、虾细菌感染和海参的疾病[1]。然而,用药不规范对渔业生产和生态环境都有严重的潜在危害,也会导致药物在奶和动物、水产品组织中的残留,使人体产生耐药性导致免疫力降低,也会使过敏个体存在潜在的风险,从而危害人体健康[2]。同时,抗生素残留也成为国际贸易的壁垒[3]。我国农业部235号公告规定苄星青霉素在动物源组织中的限量值为50 μg/kg、苯唑西林为300 μg/kg,欧盟指令NO.2377/90/EEC对青霉素类抗生素在动物源性食品中的残留进行了严格限制。所以对动物源性食品中此类抗生素残留的有效监测已经成为保障食品安全的一个重要环节。为满足残留检测的需要,建立一种动物源性食品中青霉素类抗生素残留的分析方法非常重要。
青霉素类药物残留的检测方法一直以来都是科研人员关注的热点,目前已报道的方法主要有微生物法[4]、免疫分析法[5-6]、近红外光谱法[7]、拉曼光谱法[8]、高效液相色谱-紫外法[9-13]、高效液相色谱-荧光法[14]、高效液相色谱-化学发光法[15]、液相色谱-质谱法[16-19]、电泳法[20]等。其中,微生物法操作快速简便、价格便宜,但专属性不足,容易产生假阳性;免疫分析法由于要选择配体发生结合反应,线性范围窄,在检测过程中也易产生假阳性;光谱法在定量方面存在缺陷;液相色谱-质谱法可避免生物法在准确性方面的欠缺,灵敏度高、选择性强,但由于仪器价格昂贵,不能满足绝大多数实验室的需求。高效液相色谱法成为这类药物残留应用最广泛的检测技术,其准确性强、灵敏度高,能满足青霉素类药物残留的痕量检测需求。
本实验旨在建立一种动物源性食品中青霉素G、苯唑西林、乙氧萘青霉素、双氯青霉素4 种青霉素类药物的高效液相色谱检测方法。采用乙腈-水提取目标化合物,HLB固相萃取柱净化,高效液相色谱法对不同基质中4 种青霉素类药物进行测定,并验证了所建方法的选择性、准确性、稳定性和灵敏性。
1 材料与方法
1.1 材料与试剂
青霉素G钾盐(纯度99.6%)、苯唑西林(纯度91.0%)、乙氧萘青霉素(纯度99.0%)、双氯青霉素(纯度97.5%)、水合氨苄青霉素(纯度98.5%)德国Dr. Ehrenstorfer Gmbh公司;乙腈、甲醇(HPLC级) 美国Fisher公司;乙酸铵(色谱纯) 北京Dikma公司;磷酸氢二钠、磷酸二氢钾(均为分析纯)国药集团化学试剂有限公司。
1.2 仪器与设备
1260 Infinity高效液相色谱仪(配有二极管阵列检测器)、C18色谱柱(150 mm×4.6 mm,5 μm)美国安捷伦公司;SCR20BA离心机 日本日立公司;RE52CS-2旋转蒸发仪 上海亚荣生化仪器厂;Oasis HLB固相萃取柱(500 mg,3 mL)、C18固相萃取柱(500 mg,3 mL) 美国Waters公司;HGC-36A氮吹仪天津市恒奥科技发展有限公司。
1.3 方法
1.3.1 标准溶液配制
标准储备液的配制:准确称取青霉素G、苯唑西林、乙氧萘青霉素和双氯青霉素的标准品各10 mg(精确到0.000 1 g)于10 mL容量瓶中,用水溶解并稀释至刻度,配制成质量浓度为1.00 mg/mL的青霉素G、苯唑西林、双氯青霉素和乙氧萘青霉素标准贮备液,于4 ℃以下避光保存。
标准工作液的配制:分别量取青霉素G、苯唑西林、双氯青霉素和乙氧萘青霉素质量浓度为1.00 mg/mL标准贮备液各1.00 mL于100 mL容量瓶中,用水溶解并稀释至刻度,配制成质量浓度为10.0 μg/mL的4 种青霉素混合标准工作液,于4 ℃以下避光保存。
内标储备液的配制:准确称取水合氨苄青霉素标准品10 mg(精确到0.000 1 g)于10.0 mL容量瓶中,用水溶解并稀释至刻度,配制成质量浓度为1.00 mg/mL的水合氨苄青霉素标准贮备液,于4 ℃以下避光保存。
内标工作液的配制:量取1.00 mL 1.00 mg/mL水合氨苄青霉素标准贮备液于100 mL容量瓶中,用水溶解并稀释至刻度,配制成质量浓度为10.0 μg/mL的水合氨苄青霉素标准工作液。
1.3.2 高效液相色谱条件
C18色谱柱(150 mm×4.6 mm,5 μm);流动相A为0.02 mol/L磷酸二氢钾溶液,流动相B为乙腈。梯度洗脱程序:0.00~3.00 min,87% A;3.01~10.0 min,87%~65% A;10.01~15 min,65%~87% A。流速1.0 mL/min;进样量20 μL;柱温30 ℃;紫外检测波长210 nm。
1.3.3 样品前处理
提取:准确称取切碎均质良好的样品2.00 g于50 mL离心管中,加内标工作液20 μL,静置10 min,先加入2 mL水,再加入20 mL乙腈,涡旋混匀30 s,振荡提取15 min,12 000 r/min离心5 min,将上清液全部转移至旋蒸瓶中,向残渣中再加水1 mL、乙腈10 mL涡旋混匀,二次提取,将上清液全部转移到旋蒸瓶中。在旋蒸瓶中加入0.1 mL饱和食盐水,于45 ℃水浴旋蒸至近干,加入3 mL正己烷,涡旋混匀,除去上层正己烷,剩余液体为固相萃取上样液。
换热站供热节能改造工程一般包括以下内容:(1)加装或更换一次网控制阀、热量表、远传水表和电表,将换热站内热、水、电的能源消耗情况进行独立的统计,建立能耗统计和分析系统,监测换热站的能耗情况,并制定运行调整方案。(2)二次管网加装电动或手动平衡阀,通过其调节性能,提高二次管网的输配效率,同时消除管网末端供暖质量问题,减少因供暖质量引起的人为放水。(3)站内水泵变频和PLC控制柜改造,完善站内自动控制系统,增加水泵、阀门的运行调节方式,加装或利用站内原有温度、压力测点进行数据采集。实现一次网阀门多控制方式选择,同时满足上位系统调整需求。典型换热站节能改造示意图如图1所示。
净化:将上样液转移到HLB固相萃取柱中(使用前用3 mL甲醇、3 mL水和3 mL pH 8.5的磷酸盐缓冲液活化),再用3 mL磷酸盐缓冲液洗涤鸡心瓶2 次,洗液一并转移到萃取柱中;再用3 mL水淋洗并抽干萃取柱,用2 mL甲醇+2 mL乙腈洗脱并收集于5 mL带刻度的玻璃瓶中,50 ℃氮吹至小于1 mL,用乙腈定容至1 mL,涡旋混合,过0.22 μm滤膜,供高效液相色谱分析。对每一个待测样品平行测定3 次,取平均值。
1.4 数据处理
采用SPSS 16.0统计软件进行数据统计分析,采用单因素方差分析(ANOVA),数据以 ±s表示,P<0.05,表示差异显著。
2 结果与分析
2.1 仪器条件的优化
由于青霉素类化合物在弱酸性条件下易水解,水解产物中含有巯基、羰基等,易与金属离子形成络合物,国标[21]和文献[22]因此采用衍生化法测定此类化合物。但会用到氯化汞等毒性物质,操作也较为复杂。而青霉素类化合物是β-内酰胺类,具有苯环、羰基等发色基团,在紫外波长范围内有吸收峰。故本实验采用直接进样法检测这类化合物。
青霉素类药物由于具有不稳定的四元环,易发生反应。用外标法测定时,回收率较低,故本实验选取性质接近的水合氨苄青霉素作为内标物用以提高回收率。
本实验在大量文献和前期工作的基础上,对比乙腈-水、乙腈-0.02 mol/L乙酸铵溶液、乙腈-0.02 mol/L磷酸二氢钾溶液和乙腈-0.05 mol/L磷酸二氢钾溶液作为流动相,标准物质出峰的半峰宽、分离度、保留因子等参数情况。结果表明:采用乙腈-水为流动相进行实验时,溶剂峰严重拖尾,且化合物不出峰,原因可能是流动相中不含盐,与固定相无保留;在流动相中加入盐后,化合物与固定相作用,可以正常出峰,但乙腈-0.02 mol/L乙酸铵溶液为流动相时,色谱峰的半峰宽大于乙腈-0.02 mol/L磷酸二氢钾溶液为流动相时的峰宽;加大盐的浓度,峰形变窄,变尖。但考虑到盐浓度增大在色谱柱中易结晶,影响色谱柱寿命,本实验选取乙腈-0.02 mol/L磷酸二氢钾溶液作为流动相。
以不同体积比的0.02 mol/L磷酸二氢钾-乙腈溶液作为流动相,考察4 种青霉素混合标准溶液的出峰时间。流动相比例为0.02 mol/L磷酸二氢钾-乙腈(70∶30,V/V)溶液等度洗脱,在12 min内可完成分离,如图1A所示。但内标峰水合氨苄青霉素在2.6 min左右出峰,青霉素G出峰时间在3.8 min左右,出峰时间较早。因动物源食品成分复杂,可能会对水合氨苄青霉素、青霉素G的分析产生干扰。因此本实验选取梯度洗脱,洗脱程序如1.3节所述,分离效果图见图1B。出峰顺序依次为水合氨苄青霉素、青霉素G、苯唑西林、乙氧萘青霉素和双氯青霉素。
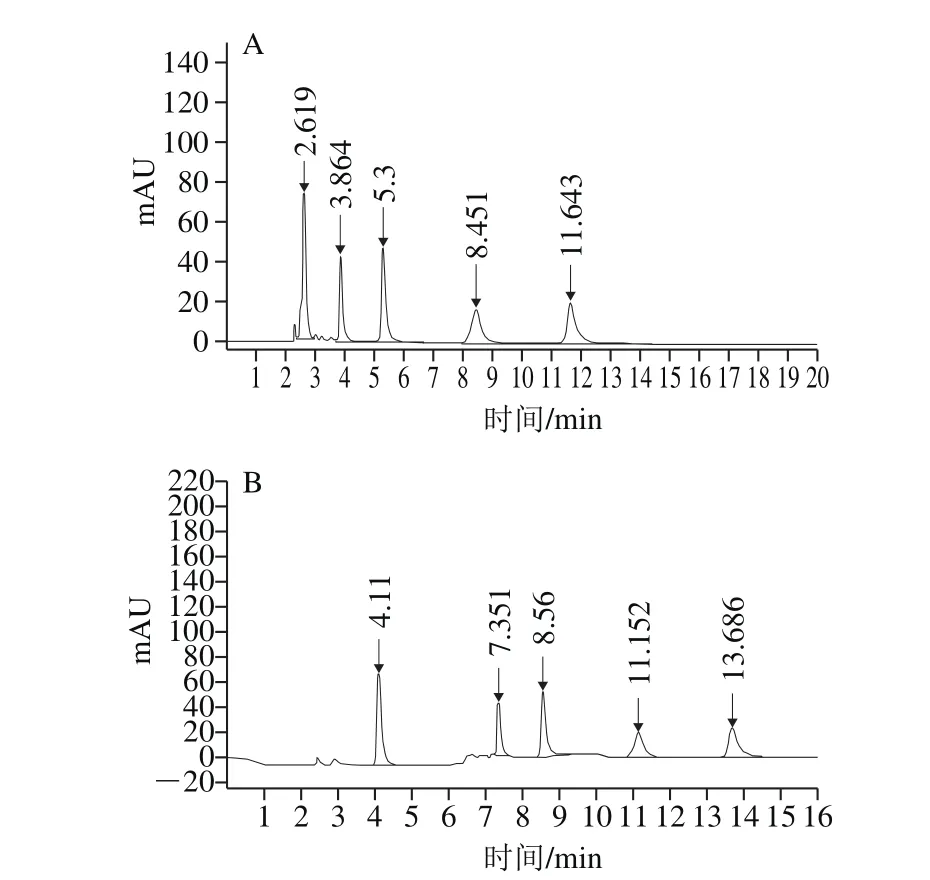
图1 等度洗脱(A)和梯度洗脱(B)色谱图Fig. 1 Chromatograms obtained by isocratic elution (A) and gradient elution (B)
2.2 提取条件的选取
2.2.1 提取溶剂的选择
参照文献方法,本实验比较了乙腈[23]、乙腈-水(10∶1,V/V)[24]、乙腈-水(4∶1,V/V)[19]、丙酮-水(1∶1,V/V)[25]作提取溶剂,其他条件如1.3.3节所述,加标量在50 μg/kg时的回收率,结果见图2。用乙腈和乙腈-水(10∶1,V/V)作提取溶剂时,回收率均在90%左右,后者回收率略高,其他2 组回收率较低。可能是由于乙腈提取时沉淀蛋白完全,易于离心分离,提前加入水能先溶解目标化合物。故本实验选取乙腈-水(10∶1,V/V)作为提取溶剂。减压蒸馏过程中加入饱和食盐水,可以防止乙腈在旋蒸过程中暴沸[24]。
2.2.2 SPE柱的选择
由于动物源性食品基质复杂,选用SPE净化小柱获得较好的净化效果。青霉素类化合物分子中含有羟基,显酸性,易溶于甲醇、乙醇、乙腈等有机溶剂。文献中广泛采用的是。本实验比较了这2 种SPE柱在相同条件下的回收率。加标量为50 μg/kg,3 mL甲醇、3 mL水活化,上样,水淋洗后抽干,最后用2 mL甲醇+2 mL乙腈洗脱,N2吹干后乙腈复溶。如图3所示,HLB柱对目标化合物的回收率均高于C18柱,与文献[24-25]报道结果一致。
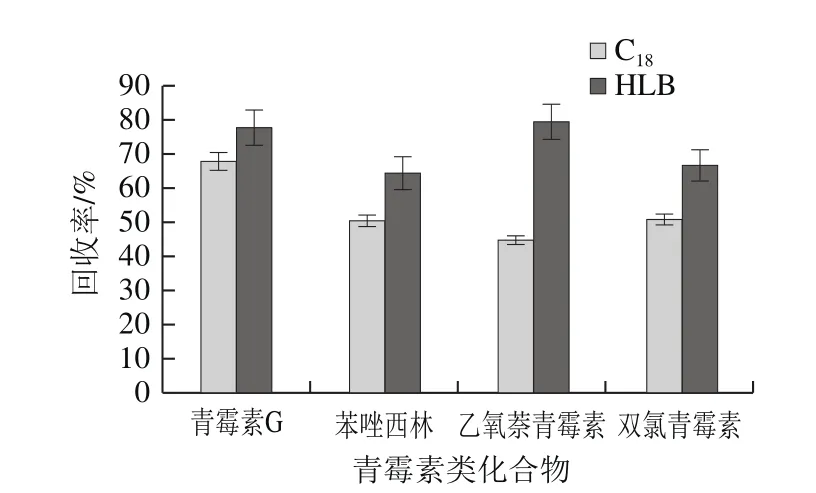
图3 不同SPE柱的回收率Fig. 3 Effect of different SPEs on recovery of penicillins
2.2.3 SPE洗脱条件的优化
不同的淋洗和洗脱溶剂也影响了SPE柱的净化效果。本实验在文献的基础上,比较不同的淋洗和洗脱溶剂对回收率的影响,如表1所示。
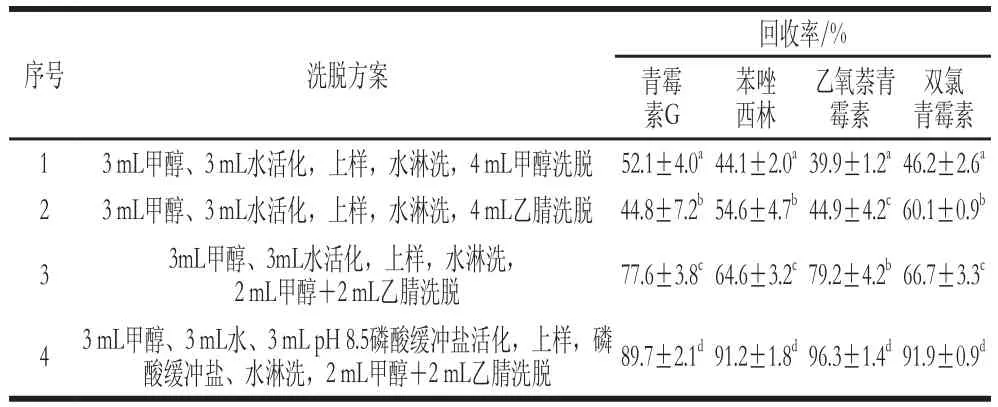
表1 SPE洗脱条件的优化Table 1 Optimization of SPE conditions
从表1可以看出,洗脱液为2 mL甲醇+2 mL乙腈时,4 种化合物的回收率明显提高。当在活化和淋洗剂中加入碱性磷酸缓冲盐时,回收率达到了90%左右。可能是碱性磷酸盐缓冲液防止待测物降解[12]。最终确定SPE萃取小柱的最佳条件为:选取HLB萃取柱,采用方案4的方式净化。
2.3 方法的验证
2.3.1 标准曲线、检出限和定量限结果
配制质量浓度分别为0.01、0.05、0.10、0.50、1.00、2.00 μg/mL 6 个水平4 种青霉素的标准混合溶液,含内标0.20 μg/mL,待高效液相色谱分析。每个溶液分别进样3 次,以目标化合物与内标质量浓度之比对其峰面积之比绘制4 种青霉素标准曲线,其中X为青霉素与内标物的质量浓度比,Y为二者峰面积之比。从表2可以看出,各目标化合物标准曲线的相关系数均大于0.999,显示了良好的相关性。检出限和定量限采用空白样品中添加青霉素类化合物的方法,分别以信噪比不小于3和信噪比不小于10计算。从表2可以看出,各青霉素的检出限为3 μg/kg,定量限为10 μg/kg,可以满足青霉素类化合物的定性及定量测定要求。

表2 4 种青霉素的标准曲线及其相关系数、检出限和定量限Table 2 Linear ranges, calibration curves, LODs and LOQs for four penicillins
2.3.2 精密度测定结果
在空白猪肉样品中进行精密度实验,样品添加不同含量的标准溶液后,涡旋混匀,于室温放置10 min,使待测成分与样品机体成分互相作用达到平衡,按1.3.2节和1.3.3节所述方法,做6 次重复实验,测定结果的相对标准偏差在10%以内。
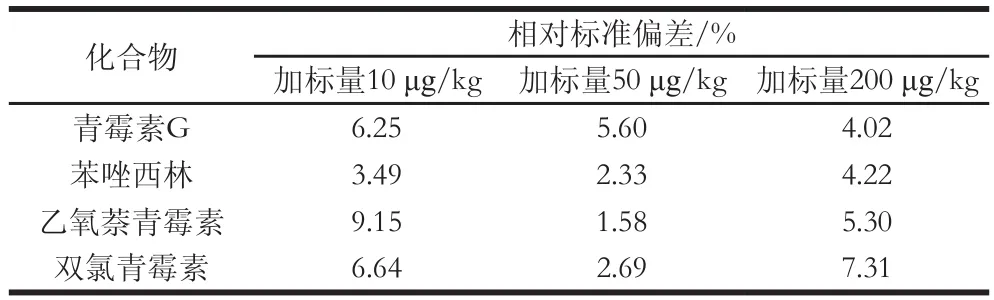
表3 精密度实验(n=6)Table 3 Results of precision test (n=6)
2.3.3 回收率测定结果
在空白样品中分别加入低、中、高3 个质量浓度的标准品进行回收率计算,选择分别加入10、50、200 μg/kg 4 种青霉素混合标准工作液,利用上述确定的检测方法,每个加标量做3 次平行实验。由表4可知,在不同的基质中,3 个水平的回收率在69.9%~100.3%范围内。从精密度和回收率的结果可以得出,该方法有较高的准确性和可靠性。
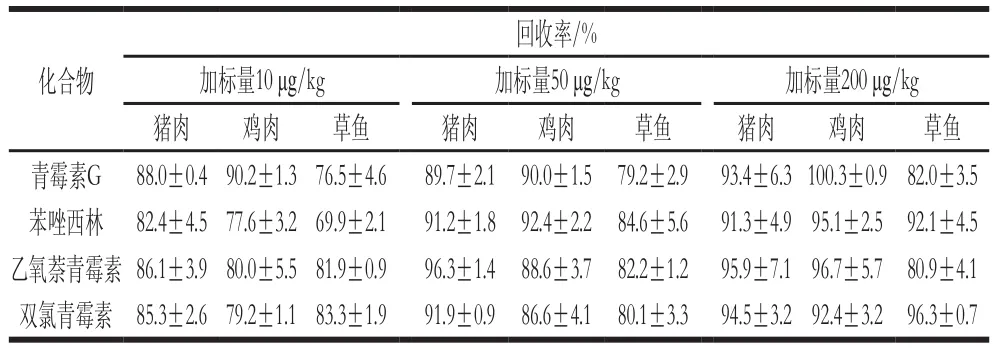
表4 4 种青霉素低、中、高3 个水平的回收率Table 4 Recovery rates for four penicillins at three different spiked levels
2.3.4 实际样品验证
从市场中选取5 种基质进行方法验证,分别为猪肉、牛肉、羊肉、鸡肉、草鱼,目标化合物出峰处均无干扰、未检测出4 种青霉素类药物残留。
3 结 论
本实验以水合氨苄青霉素作为内标,建立一种高效液相色谱法检测动物源性食品中的青霉素G钾盐、苯唑西林、乙氧萘青霉素、双氯青霉素4 种青霉素类药物残留量的方法。采用直接进样,无需衍生化,内标法定量提高了回收率。该方法前处理简单、快速、准确、灵敏,可以满足实际检测分析的需要。
