CO2钙基吸附剂在急速加热下释放机理的研究*
2019-01-03郭常青谭弘毅刘光华闫常峰
彭 垚,郭常青,谭弘毅,刘光华,闫常峰†
(1.中国科学院广州能源研究所,广州 510640;2.中国科学院大学,北京 100049;3.百大新能源股份有限公司,广东 东莞 523808)
0 引 言
化石燃料的大量使用产生的CO2等温室气体随意排放,造成的全球变暖严重影响到了地球的生态环境和全球气候[1-2]。研究CO2减排对全球各国具有紧迫而重要的意义。
利用CaO与CO2反应吸附脱除CO2以及CaCO3分解为CaO与CO2的钙循环过程,不仅普遍适用于烟气脱碳(如燃烧后钙基循环脱碳[3]),还可用于化学化工过程(如乙酸重整反应吸附强化制氢[4-5])。CaO不仅具有较高的CO2摩尔吸附容量(理论上为0.786 g CO2/gCaO),而且广泛存在于天然石灰石中,其脱碳成本较低,具有极大的经济优势[6]。因此充分了解CaO基CO2吸附剂的吸附与释放机理,对提高CO2吸附剂重复循环使用,降低钙循环脱碳技术成本,具有重要的科研价值与实际意义[7]。
目前,国内外对CaO基吸附剂的研究主要集中在CaO反应吸附CO2过程[8-11],而CaO循环利用中CaCO3分解反应的研究中,对粒径[12-13]、纯度[14-15]、加热速率[16-19]、CO2分压[20]以及颗粒的破碎、烧结[21]等有比较充分的研究。对于CaCO3分解机理的研究主要是用程序控制升温下进行的热重分析方法,如GALAN 等[18]利用热重分析(Thermogravimetric analysis, TGA)研究了不同加热速率下CaCO3的分解特性,发现CaCO3的分解温度随加热速率的增大而升高,但分解速率变慢。TIAN等[22]利用TGA进行了不同加热速率下的 CaCO3分解实验,发现CaCO3分解动力学模型分为三步。FLORIN等[20]利用TGA研究了CO2分压对CaCO3分解的影响,CUI等[13]研究了CaCO3粒径与活化能的关系。但是,由于TGA本身设计的原理与结构,一方面其加热分解速率在100℃/min(1.67℃/s)以下,温度滞后严重,与实际反应过程中加热速率为102~ 105℃/s相差甚大[17],且 TGA 测试中存在严重的扩散影响[23],其测试过程并不能真实地反映实际过程,所测得参数偏离了本征反应参数。因此 TGA实验中的 CaCO3分解反应也就不能体现真实工业反应过程。
由于热重分析仪测试中存在的一些问题,本文自主设计了一套急速加热和快速质谱气固相分析仪,并进行了急速加热下CO2钙剂吸附剂释放机理的研究,弥补CaCO3实际急速升温释放机理研究的匮乏,为能源化工和环保领域的应用提供参考。
1 实验部分
1.1 实验装置
急速加热和快速质谱气固相反应分析仪系统如图 1,样品在急速加热装置内实现快速升温反应过程,在反应的同时通过飞行时间质谱分析仪获得实时的气体产物组分与浓度数据,通过热电偶实时高速采集样品的升温数据,急速加热装置在电路系统控制下实现不同的升温速率和反应终温要求。
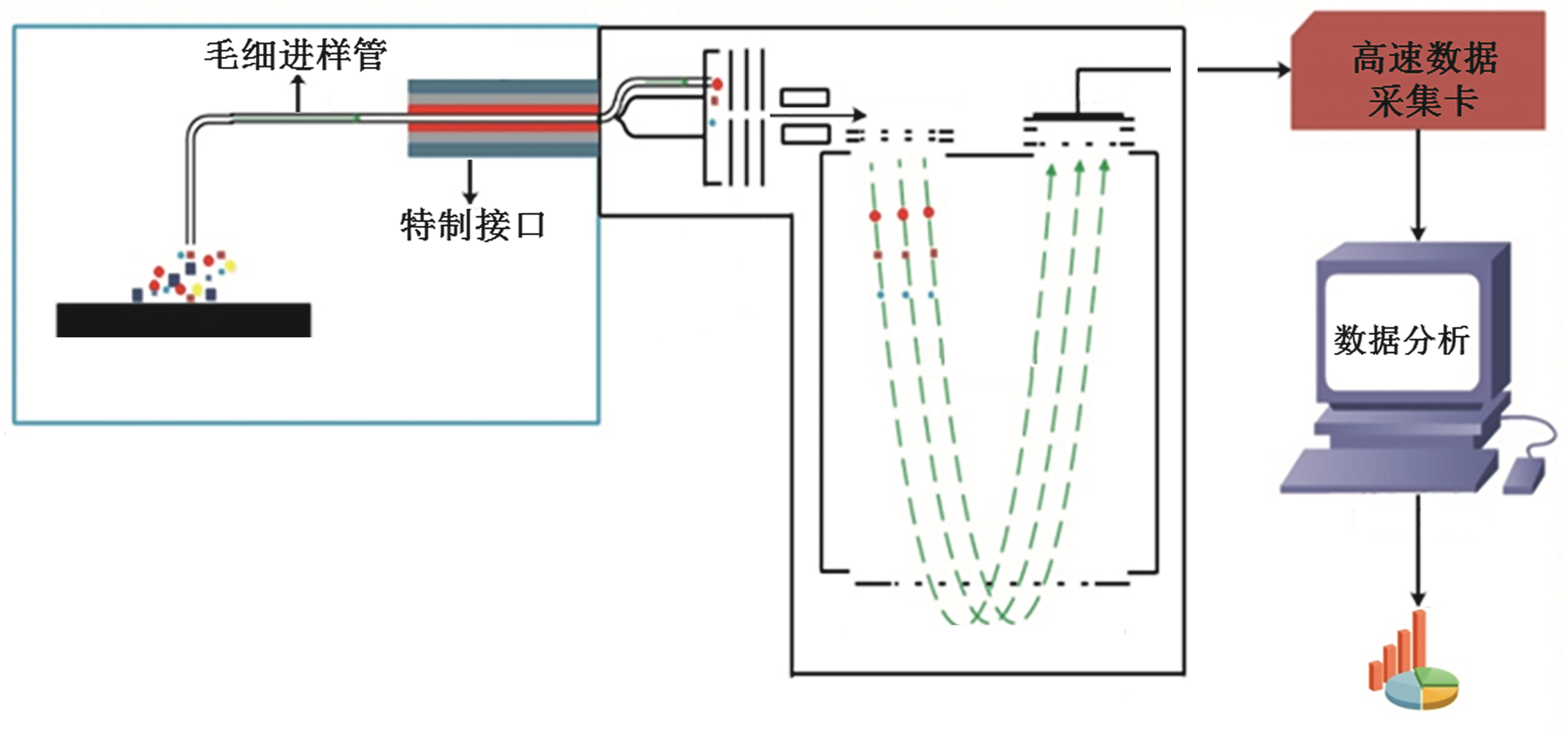
图1 急速加热和快速质谱气固相分析仪Fig.1 Gas-solid analyzer of fast heating with mass spectrometry
1.2 实验材料
CaCO3(分析纯),天津市大茂化学试剂厂;高纯氮气(纯度99.999%),广州盛盈气体有限公司;飞行时间质谱仪SPIMS-1000,广州禾信分析仪器有限公司。
1.3 实验方案
实验中筛选平均粒径为60 μm的CaCO3试剂,称取10 mg均匀置于急速加热装置中央位置,在不同的加热速率下进行分解实验,由热电偶和质谱仪实时采集 CaCO3颗粒的升温数据和 CO2的生成数据,采用高斯平滑去除仪器信噪[24]。
2 结果与讨论
2.1 不同加热速率下CaCO3分解
为模拟CaO基CO2吸附剂实际分解反应过程,建立准确的预测模型,须获得升温过程中颗粒加热速率高达 1 × 102~ 1 × 105℃/s的反应速率,但过高的升温速率又导致CaCO3分解太快而无法精准地捕捉反应过程信息[17],故本文选择 1 × 102~ 1 × 103℃/s升温速率段进行分解反应动力学特性研究。图2a ~图 2d是最大升温速率为 300℃/s、500℃/s、600℃/s、800℃/s加热速率下CaCO3颗粒的温度曲线、加热速率曲线和CO2生成曲线。图3为CO2生成量M随时间t和温度T的变化情况。

图2 不同加热速率下CaCO3分解曲线图Fig.2 CaCO3 decomposition curve at different heating rates

图3 不同加热速率下M-t图(a)和M-T图(b)Fig.3 M-t (a) and M-T (b) at different heating rates
2.2 分解特性分析
为了便于分析,如图4所示在典型分解曲线上进行相应的标记。加热起始时间点为t= 0 s,质谱仪信号值第一次大于10-11mol时间点为t1,对应温度曲线上温度值为T0,质谱仪信号峰值时间点为t2,峰值大小为Mm,对应温度为Tm,质谱仪信号值最后一次大于 10-11mol时间点为t3,t0、t1、t2、t3之间的时间间隔分别为Δt1、Δt2、Δt3。将不同加热速率下的各参数对比汇总于表1。

图4 典型CaCO3分解Fig.4 Typical CaCO3 decomposition
从图3和表1可以看出,随着加热速率的增大,Δt1、Δt2、Δt3均减小,但T0、Tm与Mm均增大,这表明加热速率的增大会使CaCO3的分解速率加快和分解温度升高,使CaCO3分解在时间尺度上提前,在温度尺度上滞后,这与GALAN等[18]在TGA实验中的发现一致,加热速率的增大可能导致反应温度的滞后,从而CaCO3热分解的温度范围变宽,分解温度的升高。

表1 不同加热速率下参数对比Table 1 Comparison of parameters at different heating rates
2.3 转化率与反应速率
CaCO3分解时转化率α由下式求出:

式中,F为气体总流量;CCO2为质谱仪采集.到的气体中CO2的体积分数,MrCaCO3为CaCO3的相对分子质量,Vm为标况下的气体摩尔体积,mCaCO3为CaCO3样品质量。

图5 CaCO3在不同加热速率下的转化率(α)Fig.5 Conversion (α) of CaCO3 at different heating rates
图5a为不同加热速率下CaCO3的转化率α随时间t的变化情况,其中小图是同一样品在TGA中100℃/min和50℃/min的测量结果。从图5a可以看出,在急速加热器内CaCO3完全反应仅需8 s,而在TGA测试中则需900 s,由此可知急速加热器反应速率明显快于同条件下的TGA。图5b是转化率α随温度T的变化情况,随升温速率的增大,CaCO3完成分解的温度升高,起始转化温度升高。样品的瞬时加热出现快速反应阶段,使得反应速率在初始阶段随转化率的增大而增快直到最大值,揭示了其急速升温特性,可见加热速率的提升有利于加快CaCO3整体反应过程的反应速率。
2.4 最可几动力学模型函数判定
气固反应动力学研究的关键是建立合理的反应机理函数模型以加深对分解过程的认识。一般气固反应速率描述方程的基本形式如下:

其中,f(α)为反应动力学方程,k(T)是与温度相关的反应速率;常采用的是阿尼乌斯公式(3),E为活化能,A为指前因子,R为通用气体常数[25]。在非等温动力学反应研究中,将温度T随时间t变化的升温速率β=T t代入式(1)即可得到非等温动力学方程:

非等温反应动力学方程积分式G(α)如下:
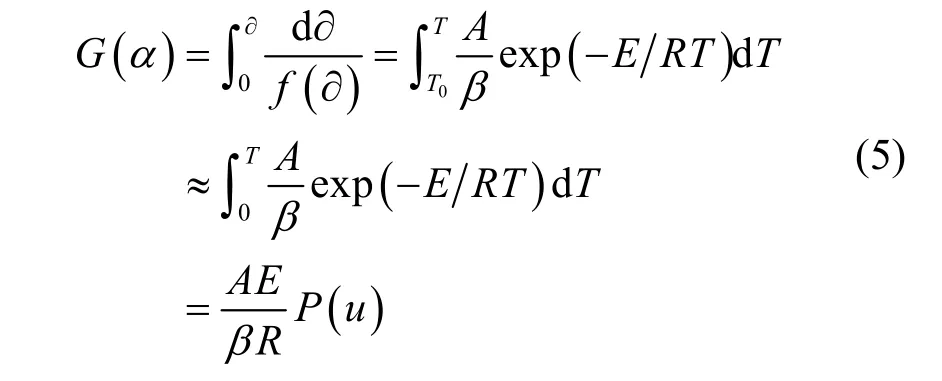
式中,一般初始反应温度较低,反应速率可忽略不计,积分下限T0的积分值趋近于 0[25],其中u=ERT,温度积分式P(u)的表达式为:

CaCO3热分解作为典型的气固反应,其动力学分析在很多文献都有报道[12-20]。本文根据不同的分解机制选取了五种常用的分解模型如表2所示。

表2 部分常用的反应动力学模型Table 2 Several common kinetic model functions for the present analysis
为了准确描述反应动力学过程,最常用的方法是通过多升温速率等转化率求得的活化能与几个可能的动力学模型的结果进行比较,从而确定最可几动力学模型函数。
(1)转化率α与活化能E的关系
采用非等温等转化率Ozawa-Flynn-Wall(OFW)方法公式如下:

从图5b上截取不同升温速率β下相同转化率α时温度T的值,由lnβ对1/T作一次线性拟合,由斜率可得在该转化率α时活化能E的数值。如图 6所示,不同转化率下曲线线性度良好。从图7可以看出,急速加热下的 CaCO3热解活化能变化范围在 113 ~ 156 kJ/mol,平均活化能为 129.38 kJ/mol,明显小于同条件下 CHEN 等[24]和谭广雷等[26]的TGA测量结果,可知急速加热装置内反应速率明显快于TGA,能有效抑制扩散作用影响,使得分解反应更加趋向于本征反应过程。且活化能随转化率的增大而降低,这与HU等[12]和GALAN等[18]在TGA实验中的发现一致,根据弗兰克尔形核动力学理论,升温速率越快,加热相同时间的过热度越大,形成临界核的半径越小,因此临界活化能会降低。
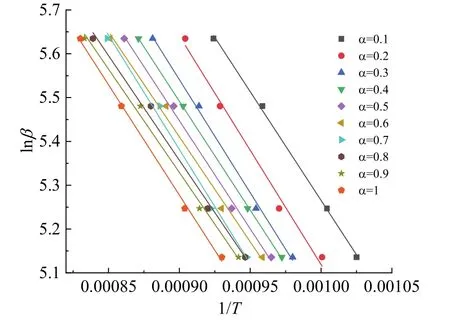
图6 lnβ-1/T拟合曲线图Fig.6 Fitting curves of lnβ versus 1/T

图7 活化能随转化率变化图Fig.7 Change of activation energy with conversion
(2)“主曲线法”判定最可几动力学模型函数
在积分式反应动力学方程(5)中,以0.5α=为参考点,可得:

采用Senum-Yang四级温度积分近似式(9)代替温度积分P(u)[27]:

将所得活化能E的平均值和各转化率α处温度T的值带入式(8),构成实验曲线P(u)P(u0.5),将表2中各种可能的动力学模型函数作G(α)G(0.5)构成标准曲线。

图8 动力学模型实验曲线与标准曲线对比图Fig.8 Comparative between experimental curve and standard curve of kinetic model
如图8所示,实验曲线不与所列出的任何一个标准曲线重叠,但实验曲线的趋势和标准曲线AE_ 3 5和AE_ 1 2的趋势相同,判定该类动力学模型函数的动力学指数经过适当修正后可以描述该热分解反应动力学机理,即:

(3)动力学指数n
将“主曲线法”判定的最可几动力学模型函数式(10)带入非等温动力学方程式(4)并取自然对数可得式(11):

在不同α和T处以 ln ( 1 -α) [ - l n ( 1-α)]、1T对ln( dαdT)作二元线性回归分析,计算可得:n= 2/5、A =806 129 s-1。
因此,根据最可几动力学模型函数判定,CaCO3在急速加热下的动力学模型函数为:
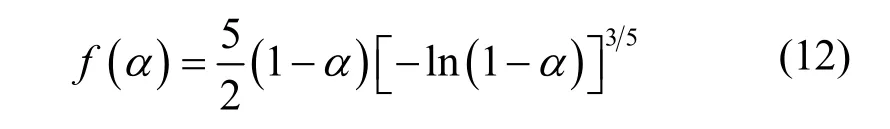
(4)重构标准曲线
将所得的动力学模型函数式(12)带回“主曲线法”方程式(9)即可得重构的标准曲线如图9所示,可以看出计算所得的动力学模型函数与实验拟合较好。

图9 重构标准曲线图Fig.9 Reconstructing standard curves of kinetic model
3 结 论
为了研究高加热速率下的钙基CO2吸附剂释放机理,本文利用自主开发的急速加热和快速质谱气固相分析仪进行了 CaCO3在 N2氛围下 300℃/s、500℃/s、600℃/s和800℃/s下的分解反应研究,结果发现:
(1)在急速加热条件下的反应速度快于一般气固反应装置,加热速率的提高使得CaCO3热分解温度升高和分解速率均加快。

(3)急速加热下的CaCO3分解反应速率比TGA快,活化能小于同条件下TGA测得的活化能,且动力学机理符合随机成核及长大模型,与TGA等慢速加热实验中测得的收缩核模型存在较大差异。
