身材矮小合并短指(趾)畸形3例家系的基因突变与表型分析并文献复习
2018-12-24胡旭昀李孟婷陈佳佳李晓侨陈少科沈亦平巩纯秀
胡旭昀 吴 迪 李孟婷 陈佳佳 李晓侨 苏 畅 陈少科 沈亦平,4,5 巩纯秀*
(1.国家儿童医学中心 首都医科大学附属北京儿童医院-北京市儿科研究所医学遗传中心 儿科重大疾病研究教育部重点实验室 出生缺陷遗传学研究北京市重点实验室, 北京 100045; 2.国家儿童医学中心 首都医科大学附属北京儿童医院内分泌遗传代谢科 儿科重大疾病研究教育部重点实验室 出生缺陷遗传学研究北京市重点实验室, 北京 100045; 3.广西壮族自治区妇幼保健院遗传代谢中心实验室,南宁 530003; 4.上海交通大学医学院附属上海儿童医学中心医学遗传科(分子诊断实验室), 上海 200127; 5.哈佛医学院波士顿儿童医院遗传科, 美国波士顿 02115)
短指(趾)畸形(brachydactyly,BD)形容指(趾)相比其他长骨或身体其他部位在比例上相对较短,是一种由于指(趾)骨、掌(跖)骨发育异常导致指(趾)缩短的疾病。BD可作为一种独立的疾病出现(多为显性遗传),也可伴随其他畸形作为复杂综合征(如Robinow综合征、Rubinstein-Taybi综合征等)出现[1-2]。在本研究中,对来自3个不同家系的BD患者进行二代测序并分别鉴定出IHH基因、ROR2基因、PTHLH基因的致病性变异。3种基因的突变都较为罕见,本研究中ROR2基因变异c.2625dupC和PTHLH基因变异c.413delA丰富了BD患者基因突变谱。而IHH基因变异c.283_285delGAG与国外报道[3]家系突变位点一致,可作为BDA1型患者热点突变进行筛查。
1 对象与方法
1.1 研究对象
本研究3例患者均来自首都医科大学附属北京儿童医院,于2015年10月至2016年8月以身材矮小就诊于儿科内分泌门诊。本研究经首都医科大学附属北京儿童医院伦理委员会批准。
1.1.1 家系1
家系1中,先证者,女,5岁,汉族,入院前2年家长发现患儿身高较同龄同性别儿童身材矮小就诊于首都医科大学附属北京儿童医院门诊。患儿为第1胎第1产,患儿母亲孕期体健,否认围产期窒息史。足月顺产,出生体质量4 050 g,出生身长50 cm。大运动里程碑发育同正常儿童。智力发育正常。
父亲年龄31岁,身高158 cm,手、足指/趾短、宽,与患儿手足的临床表现类似。无青春期身高猛长。患儿母亲年龄31岁,身高157 cm,16岁月经初潮。
体格检查:患儿体质量12.5 kg,身高98.5 cm,身高位于同年龄同性别儿童身高标准曲线第3百分位线以下,上部量:53 cm,下部量:45.5 cm,上部量/下部量=1.16∶1。指间距92.5 cm。手、足短宽,拇指/趾短、宽,第4、5手指弯曲,远端指间皮肤皱褶缺失,中节和末节指骨短(图1)。
实验室检查:血常规、尿常规、生物化学检查(血电解质,肝、肾功能,心肌酶,血脂等)、甲状腺功能、皮质醇、促肾上腺皮质激素(adrenocorticotropic hormone,ACTH)、糖化血红蛋白、性激素均正常。生长激素激发试验结果见表1。胰岛素样生长因子-1(insulin-like growth factors 1,IGF-1)和胰岛素样生长因子结合蛋白-3(insulin-like growth factor-binding protein-3,IGF-BP3)结果见表2,均正常。性激素:促黄体生成激素(luteinizing hormone,LH) 0.14 IU/L,卵泡刺激素(follicle stimulating hormone,FSH) 0.89 IU/L,雌二醇(estradiol,E2)48.8 ng/L。垂体MRI未见异常,骨龄3岁。X线片示掌骨细但骨骺宽,近侧指骨骨骺宽,中节和末节指骨明显短小(图1 D)。

图1 家系1中先证者临床表型Fig.1 Phenotype of proband from family 1A:whole body appearance; B:hand appearance; C:foot appearance; D:X-ray of left hand.
1.1.2 家系2
家系2中,先证者,男,8岁8个月,入院前5年余(即患儿3岁时)家长发现其身高较同年龄、同性别儿童矮小,生长速度近1年长高3~4 cm。患儿为第2胎第2产,足月剖宫产娩出,出生体质量3 650 g,身长不详,新生儿期体健。大运动及智力发育正常。8个月出牙,7岁换牙。
患儿父亲36岁,身高156.1 cm,无明显青春期身高猛长,手足指趾无明显短粗。患儿母亲35岁,身高157 cm,14~15岁月经初潮。
体格检查:患儿体质量20.5 kg,身高117.5 cm,身高位于同年龄同性别儿童身高标准曲线第3百分位线以下,上部量60 cm,下部量57 cm,上部量/下部量=1.04∶1。指间距92.5 cm,拇指略宽,手足指趾骨发育无明显异常(图2)。
实验室检查:血常规、尿常规、生物化学检查(血电解质,肝、肾功能,心肌酶,血脂等)、甲状腺功能、皮质醇、ACTH、糖化血红蛋白、性激素均正常。生长激素激发试验结果见表1。IGF-1和IGF-BP3均正常(表2)。垂体MRI未见异常,骨龄5岁。X线片示指骨远端节段略短小(图2D)。
1.1.3 家系3
家系3中,先证者,女,5岁,入院前5年(即患儿生后)家长发现患儿身长较同龄同性别儿童矮。入院前4年(即患儿1岁1个月时)查身高66 cm,体质量6.9 kg,未予特殊治疗,嘱随诊观察,监测患儿生长发育情况。近1年半患儿身高增长约11 cm。患儿为第2胎第2产,孕足月剖宫产,出生体质量1 800 g,出生身长不详。大运动里程碑发育同正常儿童。智力发育正常。出牙晚,1岁始出牙,6岁换牙。
患儿父亲29岁,身高170 cm,手足指趾未见异常,15岁身高猛长;患儿母亲28岁,身高161.5 cm,12岁月经初潮。家系中患儿姑姑身高168 cm,但手指略短小(图3),姑姑未行基因检测。

图2 家系2中先证者临床表型Fig.2 Phenotype of proband from family 2A:whole body appearance; B:hand appearance; C:foot appearance; D:X-ray of left hand.

图3 家系3中先证者临床表型Fig.3 Phenotype of proband from family 3A:facial appearance, the proband had thin hair, long face, arched eyebrows, tooth abnormal and microtia; B:hand appearance; C:foot appearance; D:X-ray of left hand; E:Hand appearance of the aunt of proband 3 showed mild brachydactyly.

表1 3例先证者GH-IGF1轴激素检测结果Tab.1 Results of GH-IGF1 axis hormone tests of three patients
IGF-1:insulin-like growth factors 1;IGF-BP3:insulin-like growth factor-binding protein 3;GH:growth hormone;NA:no data.
体格检查:患儿身高93.5 cm,体质量11 kg,指间距90 cm。上部量51 cm,下部量42.5 cm。上部量/下部量之比=1.2∶1。第5小指弯曲。齿列不齐,牙齿数目正常,但每个牙齿都偏小(图3 A~C)。
实验室检查:血常规、尿常规、生物化学检查(血电解质,肝、肾功能,心肌酶,血脂等)、甲状腺功能、皮质醇、ACTH、糖化血红蛋白、性激素均正常。生长激素激发试验结果见表1。IGF-1和IGF-BP3均正常(表2)。垂体MRI:未见异常。骨龄:4岁。X线片示指骨短粗,尤第5小指中段指节短(图3D)。
1.2 遗传学检测
经家系成员书面知情同意后,抽取先证者及其父母静脉血各2 mL,使用德国Qiagen公司QIAamp Blood DNA Mini Kit对核心家系基因组DNA进行提取。采用美国Agilent公司SureSelect全外显子基因检测试剂盒对核心家系成员全外显子组进行捕获,构建文库在美国Illumina公司的Hiseq X Ten测序仪上进行高通量测序,通过TGex(tgex-app.genecards.cn)软件对测序结果进行注释和过滤。通过美国医学遗传学和基因组学协会/分子病理学协会(American College of Medical Genetics and Genomics and Association for Molecular Pathology,ACMG/AMP)指南[4]对变异进行致病性分析。
2 结果
2.1 临床随访
来自3个受累家系的先证者分别接受生长激素治疗30个月、12个月和34.5个月,身高SDS均得到明显改善,经过治疗后身高均已达到或接近正常水平。具体治疗结果见表2。
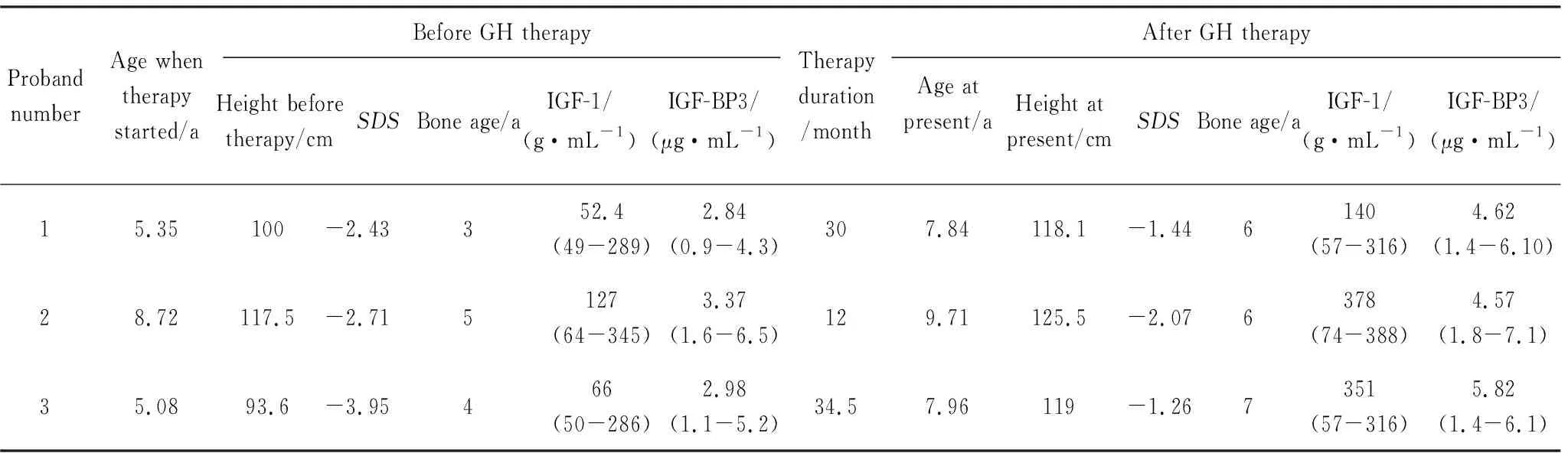
表2 3例先证者接受生长激素治疗情况Tab.2 Growth hormone treatment of three probands
IGF-1:insulin-like growth factors-1;IGF-BP3:insulin-like growth factor-binding protein 3;GH:growth hormone;SDS:standard deviation score.
2.2 遗传学诊断
经过二代测序分析,在家系1先证者中鉴定出一个遗传自父亲的IHH基因框内缺失变异c.283_285delGAG(p.E95del,图4),导致蛋白产物第95位谷氨酸残基缺失。该变异位于1号外显子,N端信号结构域区域(N-terminal signaling domain),在脊椎动物中高度保守(图5)。在家系2中鉴定出一个遗传自父亲的ROR2基因移码变异c.2625dupC(p.T876fs*20),位于9号外显子,导致变异后第20位提前出现终止密码子。在家系3中鉴定出遗传自父亲的PTHLH基因移码变异c.413delA(p.K138fs*11),位于4号外显子,导致变异后第11位提前出现终止密码子。

图5 家系1先证者中鉴定出的IHH基因变异Fig.5 IHH variant identified from Family 1A:crystal structure of the Indian Hedgehog N-terminal signalling domain and the location of p.E95; B:p.E95 highly conserved in vertebrates.
3 讨论
本研究中3例患儿均以矮小为主诉就诊内分泌科,其中2例父亲身高<160 cm,均可诊断家族性矮小。病例1在临床仔细查体时发现患儿有手指短特殊体征,患儿父亲亦有相同体征。病例2、病例3患儿临床短指表现不明显,仅影像学显示指骨短粗,故未明确诊断,利用二代测序技术,鉴定出3例患儿均为短指/趾畸形,分别为A1、B1、E2型,分子诊断为来自IHH基因、ROR2基因、PTHLH基因的突变。此3型的共同点均为手足短、宽,指骨短,干骺端略粗,可伴有趾指弯曲、出牙晚等特点。
目前与BD相关的疾病超过450种,与BD相关的基因超过250个(hpo.jax.org/app/browse/term/HP:0001156),其中,单独出现的短指畸形根据Bell分型可以分为A~E型以及相应亚型。临床上不同亚型表型上存在重叠现象,且往往会伴随身材矮小等表型,容易被诊断为特发性矮小,常需要借助遗传学诊断。在本研究中,对来自3个不同家系的BD患者进行二代测序并分别鉴定出来自IHH基因、ROR2基因、PTHLH基因的致病性变异,分别诊断为BDA1型、B1型和E2型。
3种基因得突变都较为罕见,在世界范围内(HGMD Professional 2018.2),IHH基因仅报道了31个变异,ROR2基因仅报道了40个变异、PTHLH基因仅报道了18个变异[1-2]。本研究中ROR2基因变异c.2625dupC(p.T876fs*20)和PTHLH基因变异c.413delA(p.K138fs*11),丰富了BD患者基因突变谱。同时在中国人群中报道了IHH基因变异c.283_285delGAG(p.E95del),该变异在dbSNP数据库、千人基因组数据库以及GnomAD数据库中均未见报道,在一荷兰家系中曾有该变异的报道[3],p.E95del在此家系中与5例患者存在共分离。经过ACMG/AMP指南对变异进行致病性分析,分类为可能致病性变异。该变异与国外家系报道[3]一致,从而证明该变异可能是BDA1型的一个热点突变。对于ROR2基因和PTHLH基因的移码突变,可能导致产物RNA通过无义介导的mRNA衰减机制降解或产生截短蛋白,因而突变等位基因活性大部分丧失。这两例变异在dbSNP数据库、千人基因组数据库以及GnomAD数据库中均未见报道,提示等位基因变异人群频率极低。人类基因突变数据库(Human Gene Mutation Database,HGMD)与Clinvar数据库中均未收录该变异致病报道,经过ACMG/AMP指南[4]对变异进行致病性分析,均分类为致病性或可能致病性变异。
IHH基因位于2号染色体q35区域,编码的印度刺猬(Indian hedgehog)蛋白特异性表达在软骨板,指(趾)尖和生长板中的前肥大软骨细胞。该蛋白功能涉及软骨细胞分化、关节发育与骨形成,可在发育过程中调节骨骼的生长和骨化之间的平衡,同时可诱导甲状旁腺激素相关蛋白的表达[5]。
2001年, Gao等[6]首次揭示了IHH基因突变与BDA1型的关系,在3个独立的中国大家系中,鉴定出3个N端信号结构域区域(N-terminal signalling domain)的错义变异。BDA1型的特点是所有指(趾)的成比例缩短、指间皱褶部分缺失,并常伴随第二、三、四指(趾)弯曲,以及拇指(趾)的近端指(趾)骨宽。X线片提示中指(趾)骨缩短或缺失、远端指(趾)骨缩短、末端指(趾)关节黏连以及近侧指骨变细伴骨骺增大。第二和第五指(趾)受累最为严重。在严重受累患者中可累及整个骨骼系统,导致身材矮小。本研究家系1中患者的变异同样位于N端信号结构域区域,第95号氨基酸位置,该位置此前有出现3例不同变异的报道:p.E95K、p.E95G和p.E95del[3,6-7],表明该高度保守的谷氨酸对于IHH信号通路的重要性,该家系患者临床症状及影像学表现典型,表明该结构域区域位点变异影响功能,预测在BDA1家族中会发现更多的E95突变,可作为BDA1型患者热点突变进行筛查。
ROR2基因位于9号染色体q22.31区域,编码酪氨酸蛋白激酶跨膜受体,目前研究[8]显示其功能可能涉及软骨细胞的早期形成,是软骨和生长板发育所必需的受体,从而诱导成骨作用。2000年, Oldridge等[9]在BDB1家系中鉴定出靠近ROR2蛋白C端的2个无义变异和1个移码变异。经预测这些变异会导致表达产物酪氨酸激酶结构域后的胞内部分缺失。BDB1是短指中表型最为严重的一种,特点主要表现为2~5指(趾)的远端(包括指甲)和中端指(趾)节发育不良甚至缺失,同时出现指(趾)关节黏连、指屈曲和并指等表型。
本研究中家系2重患者所携带的移码变异为世界首报,预测将产生缺少1个下游丝氨酸/苏氨酸区域的截短蛋白,影响正常功能。目前研究[10-11]表明,BDB1突变多以无义或移码变异为主,且其中大多数变异最终导致产生截短蛋白,变异位置不同引起患者表型差异较大,其中部分表型轻微患者可仅表现为个别手指弯曲或者个别手指指甲变小/缺失等[12]。本家系患者及其父亲携带的变异位置靠近末端,这可能是其表型较轻微的原因之一。
PTHLH基因位于12号染色体p11.22区域,其编码的甲状旁腺激素相关蛋白可作为内分泌、自分泌、旁分泌和胞分泌激素,是骨发育调控所必需的,维持软骨细胞的增生、延缓软骨细胞的进一步分化[13]。2010年,Klopocki等[14]在4个散发的BDE2、身材矮小家系中鉴定出该基因的2个错义变异和2个无义变异,证明了PTHLH基因与BDE2的关系。同BDA1和BDB1不同,BDE2的患者除指(趾)骨缩短外,还可表现为掌骨和跖骨不同程度的缩短,以及出牙延迟或少牙畸形。本研究家系3中患者的变异c.413delA(p.K138fs*11)为世界上首次报道,可能导致产物RNA通过无义介导的mRNA衰减机制降解或产生一个截短蛋白。本研究报道的患者具有典型的身材矮小、短指畸形以及牙列不齐、牙齿偏小(图3)。但是携带相同变异的父亲身高却是正常的,可能是存在着修饰基因等因素而导致外显不全。目前,有报道称携带PTHLH基因突变的患者中仍有约41%(14/34)具有正常身高,具体原因仍需更多研究[14-18]。
目前,对不同亚型的BD仍然没有统一的治疗方法。极端情况下,当畸形严重影响手部正常功能时可以考虑整形手术。不同亚型的BD预后情况差异较大,最新的研究[19]成果显示IHH突变在身材矮小患儿中占有较高比例且对生长激素治疗有较好的反应,Pereda等[15]对5例IHH突变患儿进行生长激素治疗后平均身高1年内增长了0.6标准差。在本研究中,由于3个家系的先证者都是以身材矮小为主诉进行就诊,对3个患儿均进行了生长激素治疗,治疗结果显著,身高SDS均得到明显改善,为合并BD的身材矮小临床治疗提供了更多依据。
综上所述,在3例身材矮小伴BD患者中分别鉴定出来自IHH基因、ROR2基因、PTHLH基因的致病性变异,不仅明确了患者的临床诊断及其分型,同时对临床表型以及治疗情况进行详细说明,从而进一步扩展了BD患者的突变谱和表型谱,为临床诊断和遗传咨询提供了直接依据。临床上针对家族性矮小,当有临床或影像学的提示时,即使症状不典型,亦建议BD的靶向捕获基因检测以进一步明确分子病因。
