Cu(OAc)2催化查尔酮肟酯与丁炔二酸二甲酯的环化反应合成多取代吡啶
2018-12-03彭文武柳佳宁
彭文武,柳佳宁,张 泽
(安徽工程大学 生物与化学工程学院,安徽 芜湖 241000)
吡啶及其衍生物是一类非常重要的含氮杂环化合物,是有机合成中十分重要的中间体,也是无机配合物中常用的一种配体,而且还广泛存在于许多天然产物和具有生理活性的化合物中[1].例如Nicotinamide、Pigolitazao、Nifedipine、Esomeprazole、Chlorpyrifos等一些常见的药物中均含有吡啶骨架.此外,吡啶衍生物在功能性材料方面也有着十分重要的应用,例如,包含多个吡啶环的化合物B3PyPB就是一种高性能有机发光器件的重要组成部分[2].鉴于吡啶类化合物在有机合成、医药化学、材料科学等领域的重要应用以及良好的发展前景,探索新型高效的功能性吡啶衍生物的合成方法一直是合成化学家们研究的热点.
肟酯是一类重要的含氮化合物,它独特的反应特性使其成为了构建含氮杂环骨架的重要前体.近些年来,不断有关于肟酯参与的合成转化反应来高效构建功能性含氮杂环化合物的报道.Tang[3]发展了一种钯催化芳香肟C(sp2)-H的羰基化反应,从而高选择性合成出一系列苯并噁嗪酮和异吲哚酮类化合物;Bergman[4]等报道了在一价铑催化下,以廉价的亚磷酸三异丙酯为配体利用端炔和肟酯高区域选择性合成了一系列多取代吡啶;Jeganmoha[5]和Ackermann[6]相继通过钌催化下环化芳基肟类衍生物与不对称内炔的交叉偶联反应,成功合成出一系列新型多取代吡啶衍生物.Bower[7]等在Narasaka-Heck反应的基础上,在廉价铜催化下通过烯丙基肟酯的分子内Heck环化反应,成功合成出烯基取代的吡咯衍生物.随后Pierce[8]采用链烯基硫代肟酯为原料,以一价的铜盐络合物为催化剂,发展出了一种高效合成噻唑啉骨架的方法.与此同时Cui[9]等以丙二腈、醛、磺酰烯胺和肟酯为原料,通过铜催化的环化反应合成了一系列功能性吡啶和吡唑啉衍生物.Yu[10]等发展了铜催化γ,δ-不饱和肟酯的环化与叠氮化反应合成了一系列的吡咯衍生物.这些合成方法均有各自优点,但大多使用稀缺、昂贵或有毒的催化剂,反应条件相对苛刻,有些需要预制中间体或对反应底物有特异性要求从而限制了方法的普适性,所以仍存在一定的局限性,因此探索更加高效、环境友好型的合成含氮杂环衍生物方法是非常有必要的.以取代查尔酮肟酯和丁炔二酸二甲酯为原料,在醋酸铜催化条件下通过环化反应合成多取代吡啶类衍生物,该方法操作简单,所使用的催化剂廉价无毒、来源丰富,且副产物多为水、甲醇、酸等低污染化合物,能在一定程度上有效弥补传统的吡啶类衍生物合成方法中存在的缺陷,从而为这一类化合物的合成提供一条新的途径.
1 实验部分
1.1 试剂与仪器
取代苯乙酮,取代苯甲醛,氢氧化钠,盐酸羟胺,丁炔二酸二甲酯,无水亚硫酸钠,亚硫酸氢钠,分子筛,氯化亚铜,碘化亚铜,醋酸铜,五水硫酸铜,氯化铜,乙酸酐,吡啶,4-二甲氨基吡啶,溶剂(AR).
数字显微熔点测定仪,旋转蒸发仪RE-52C,手提式紫外分析仪,Avance300或500型核磁共振波谱仪,薄层层析硅胶.
1.2 取代查尔酮肟酯的制备
向50 mL圆底烧瓶中依次加入10 mmol取代查尔酮[11-13]、15 mmol盐酸羟胺、10 mmol无水硫酸钠,再向其中加入30 mL无水乙醇,回流条件下磁子搅拌反应3 h.通过薄层层析色谱法检测反应物反应完全时停止反应,过滤并旋蒸得到多取代查尔酮肟粗产品,该粗产品不需要进一步纯化即可进行下一步反应.向50 mL圆底烧瓶中依次加入10 mmol取代查尔酮肟、30 mmol乙酸酐、30 mmol吡啶、5 mmol 4-二甲氨基吡啶(DMAP),再向其中加入分子筛和30 mL二氯甲烷(DCM),室温搅拌下反应1 h,通过薄层层析色谱法检测反应物反应完全时加水停止反应,反应式如图1所示.反应结束后用乙酸乙酯和水进行萃取,并用无水硫酸镁干燥,过滤旋蒸得到的粗产品用石油醚和乙酸乙酯(石油醚∶乙酸乙酯=5∶1)重结晶,得到纯净产物即为取代查尔酮肟酯.
1.3 多取代吡啶的合成(以合成1ab为例)
向试管中依次加入1 mmol取代查尔酮肟酯(1a)、1 mmol丁炔二酸二甲酯(b)、1 mmol氯化亚铜,再向其中加入2 mL二甲亚砜(DMSO),油浴加热到120 ℃,磁子搅拌反应3 h.通过薄层层析色谱法检测反应物反应完全时停止反应,反应结束后用乙酸乙酯和水进行萃取,得到的粗产品通过硅胶柱色谱(洗脱剂:石油醚∶乙酸乙酯=10∶1)分离提纯得到纯净产物.
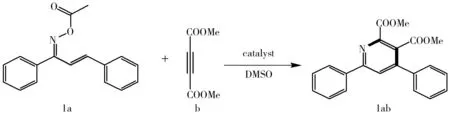
图1 查尔酮肟酯1a与丁炔二酸二甲酯b的环化反应合成多取代吡啶1ab
1.4 多取代吡啶的结构表征
1ab:4,6-Diphenyl-pyridine-2,3-dicarboxylic acid dim-ethyl ester.Yellow oil;1H NMR(400 MHz,CDCl3) δ 8.13~8.07(m,2H),7.90(s,1H),7.55~7.45(m,8H),4.05(s,3H),3.76(s,3H);13C NMR(100 MHz,CDCl3) δ 167.9,165.7,157.9,150.0,146.2,137.6,137.3,130.0,129.1,129.0,128.8,128.7,128.1,127.4,123.8,53.3,52.7.
2ab:6-Phenyl-4-p-tolyl-pyridine-2,3-dicarboxylic acid dimethyl ester.White solid;m.p.96~97 ℃;1H NMR(400 MHz,CDCl3) δ 8.14~8.06(m,2H),7.89(s,1H),7.58~7.45(m,3H),7.40~7.34(m,2H),7.32~7.24(m,2H),4.05(s,3H),3.79(s,3H),2.44(s,3H);13C NMR(100 MHz,CDCl3) δ 168.0,165.7,157.9,150.0,146.2,139.2,137.7,134.4,130.0,129.5,129.0,128.7,128.0,127.4,123.8,53.2,52.7,21.3.
3ab:4-(4-Cyano-phenyl)-6-phenyl-pyridine-2,3-dicarboxylic acid dimethyl ester.White solid;m.p.145 ℃~146 ℃;1H NMR(400 MHz,CDCl3) δ 8.18~8.02(m,2H),7.85(s,1H),7.82~7.72(m,2H),7.61~7.55(m,2H),7.55~7.46(m,3H),4.05(s,3H),3.76(s,3H);13C NMR(100 MHz,CDCl3) δ 167.2,165.5,158.3,148.1,146.8,141.9,137.1,132.5,130.4,129.1,129.0,128.0,127.4,123.1,118.2,113.1,53.4,52.9.
4ab:4-(4-Methoxy-phenyl)-6-phenyl-pyridine-2,3-dicarboxylic acid dimethyl ester.White solid;m.p.87 ℃~88 ℃;1H NMR(400 MHz,CDCl3) δ 8.15~8.03(m,2H),7.88(s,1H),7.57~7.37(m,5H),7.06~6.96(m,2H),4.04(s,3H),3.88(s,3H),3.80(s,3H);13C NMR(100 MHz,CDCl3) δ 168.2,165.8,160.4,157.8,149.6,146.1,137.8,129.9,129.5,129.5,129.0,128.7,127.4,123.7,114.3,55.4,53.2,52.8.
5ab:4-(2-Methoxy-phenyl)-6-phenyl-pyridine-2,3-dicarboxylic acid dimethyl ester.White solid;m.p.100 ℃~101 ℃;1H NMR(400 MHz,CDCl3) δ 8.06(dd,J=8.0,1.5 Hz,2H),7.83(s,1H),7.53~7.37(m,4H),7.27(dd,J=9.0,1.4 Hz,1H),7.05(t,J=7.1 Hz,1H),6.96(d,J=8.3 Hz,1H),4.00(s,3H),3.75(s,3H),3.68(s,3H);13C NMR(100 MHz,CDCl3) δ 167.2,166.4,157.8,156.0,147.9,147.6,137.8,130.5,130.0,129.9,128.9,128.1,127.4,126.4,124.5,120.8,110.8,55.4,53.0,52.3.
6ab:6-(4-Bromo-phenyl)-4-phenyl-pyridine-2,3-dicarboxylic acid dimethyl ester.White solid;m.p.103 ℃~104 ℃;1H NMR(400 MHz,CDCl3) δ 7.96(d,J=8.6 Hz,2H),7.85(s,1H),7.62(d,J=8.5 Hz,2H),7.50~7.40(m,5H),4.02(s,3H),3.73(s,3H);13C NMR(100 MHz,CDCl3) δ 167.7,165.5,156.6,150.2,146.5,146.4,137.2,136.5,132.1,129.2,128.9,128.8,128.1,124.7,123.4,53.2,52.7.
7ab:6-(4-Methoxy-phenyl)-4-phenyl-pyridine-2,3-dicarboxylic acid dimethyl ester.White solid;m.p.106 ℃~107 ℃;1H NMR(400 MHz,CDCl3) δ 8.11~8.03(m,2H),7.83(s,1H),7.48(dq,J=6.5,3.6,2.7 Hz,5H),7.06~6.99(m,2H),4.04(s,3H),3.89(s,3H),3.75(s,3H);13C NMR(100 MHz,CDCl3) δ 168.0,165.8,161.3,157.5,149.9,146.2,137.5,129.0,128.8,128.7,128.1,127.9,127.7,122.8,114.3,55.4,53.2,52.7.
2 结果与讨论
2.1 反应条件的优化
选取查尔酮肟酯(1a)和丁炔二酸二甲酯(b)合成2,3,4,6-四取代吡啶(1ab)作为模型反应,系统考察了催化剂种类、反应温度、反应溶剂、反应添加剂和反应时间对反应的影响.
反应条件的优化如表1所示.按照1a∶b∶催化剂=1∶1∶1的反应投料摩尔比,以DMSO为溶剂,反应温度为120 ℃,尝试了几种常见催化剂(结果如表1中1#~7#所示),通过薄层层析色谱法(TLC)跟踪反应发现,铜盐不管是一价还是二价均具有一定的催化效果,而在三价锰(Mn(OAc)3·3H2O)催化或不加催化剂的条件下,反应均不能发生.相比之下,二价铜盐醋酸铜(Cu(OAc)2·H2O)和氯化铜(CuCl2)的催化效果比较好,其中,醋酸铜(Cu(OAc)2·H2O)的催化效果最好.
选择最佳催化剂后,对温度进行了优化(结果如表1中8#~10#所示).结果表明,反应温度为120 ℃和70 ℃时产率均低于90 ℃和100 ℃时的产率,反应温度为120 ℃时副产物明显增多,而反应温度为70 ℃时反应不完全,综合考虑到反应的选择性和转化率,选取90 ℃为最佳反应温度.由于反应溶剂对反应有着重要影响,在上述最佳条件下,尝试了几种不同的反应溶剂(结果如表1中11#~13#所示).结果表明,以二甲基亚砜(DMSO)为反应溶剂的产率明显要高于二甲基甲酰胺(DMF)和甲苯(Toluene)的产率,这可能与DMSO的强极性和强溶剂化效应有关.另外,还尝试了在不加溶剂的条件下进行机械反应,结果发现,反应几乎不能发生.因此,综合考虑,选择二甲基亚砜(DMSO)为反应的最佳溶剂.
为进一步提高产率,尝试几种添加剂(如表1中14#~16#所示).结果表明,加入分子筛(4 Å)和无水亚硫酸钠(Na2SO3),产率几乎没有什么提高,而加入亚硫酸氢钠(NaHSO3)产率略有提高,这可能与亚硫酸氢钠能抑制肟酯的分解有关.就反应时间而言(如表1中17#~19#所示),时间缩短为1 h时反应转化率低,而2 h时转化率有所提高但低于3 h的产率,继续延长时间产率几乎没有提高,因此3 h为最佳反应时间.
综上所述,最终确定合成该化合物的最佳反应条件为:取代查尔酮肟酯∶丁炔二酸二甲酯∶醋酸铜(Cu(OAc)2·H2O)∶亚硫酸氢钠(NaHSO3)=1∶1∶1∶1,反应溶剂为二甲基亚砜(DMSO),反应温度为90 ℃,反应时间3 h.
2.2 底物的扩展
利用上述优化的最佳反应条件,以不同取代查尔酮肟酯和丁炔二酸二甲酯为原料,共合成出7种不同的2,3,4,6-四取代吡啶,底物的扩展如表2所示(1ab~7ab),并经1H-NMR及13C-NMR对每一种化合物结构进行了确认.由表2可以看出,当查尔酮骨架的苯环上取代基为给电子基团(OCH3)时,反应活性明显高一些(4ab,5ab,7ab);而当取代基为吸电子基团(CN,Br)时,反应活性明显要低一些(3ab,6ab).这与反应的机理是相符的,因为反应经历了[4+2]环加成过程,在此过程中,二烯体上有给电子基团是有利于反应进行的.
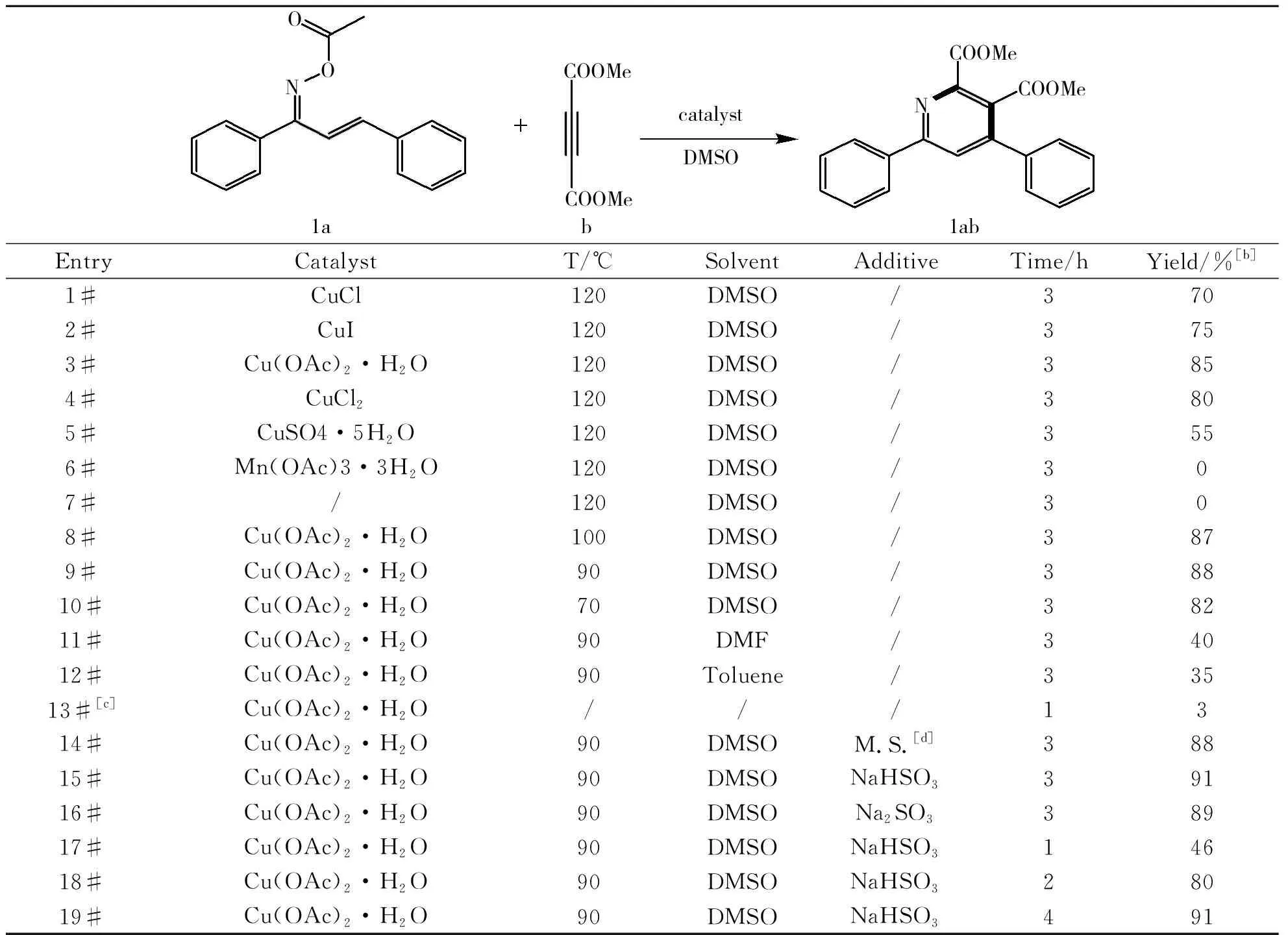
表1 反应条件的优化[a]
[a]:Reaction condition:1a(1 mmol),b(1 mmol),catalyst(1 mmol) and additive(1 mmol) were stirring in 2 mL solvent.[b]:Isolated yield.[c]:Solvent-free mechanical ball milling.[d]:M.S.=molecular sieves(4 Å).
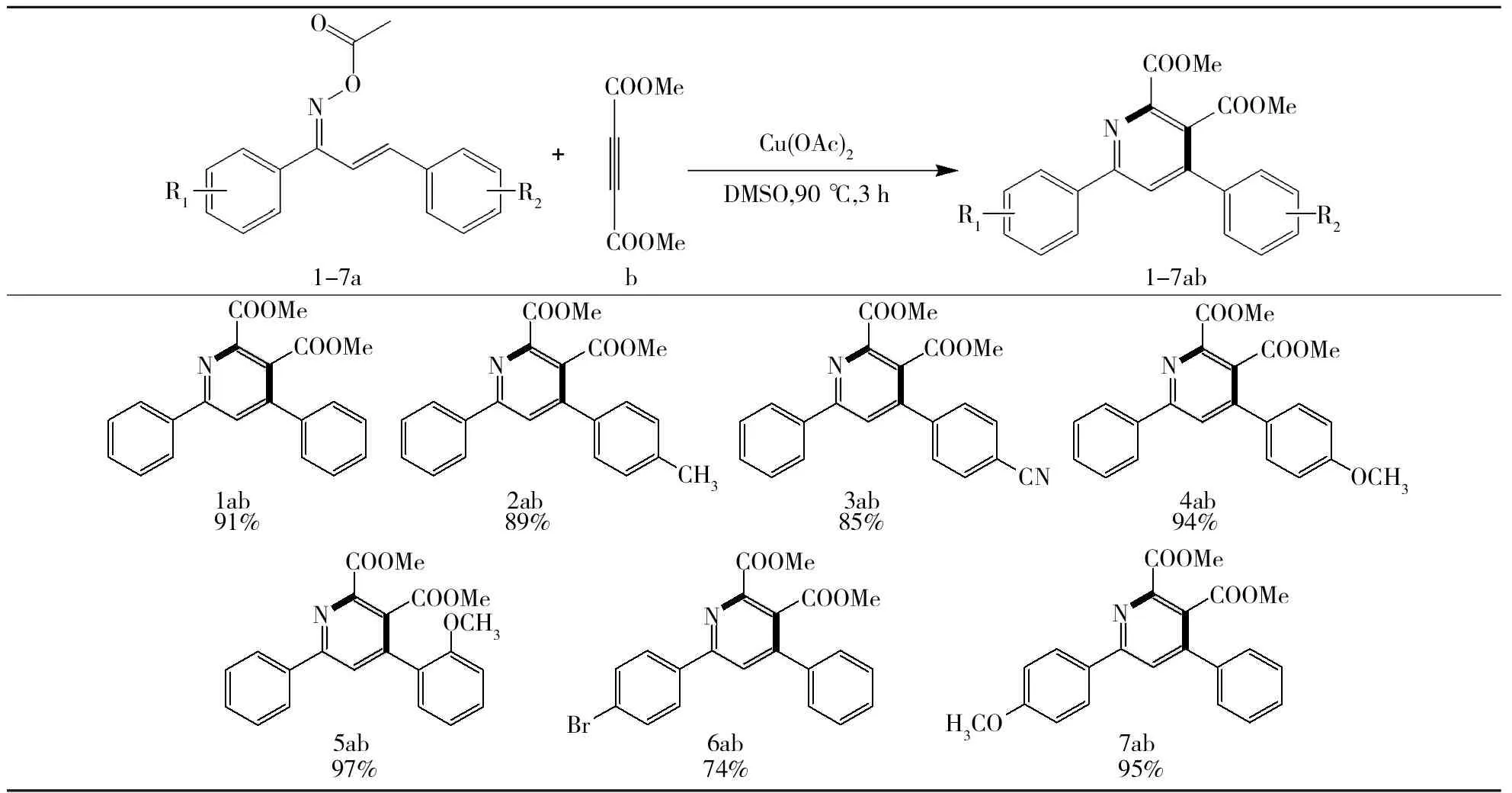
表2 底物的扩展
3 结论
以取代查尔酮肟酯∶丁炔二酸二甲酯∶醋酸铜(Cu(OAc)2)∶亚硫酸氢钠(NaHSO3)=1∶1∶1∶1为原料比例,二甲基亚砜(DMSO)作为反应溶剂,在90 ℃下反应3 h,探索发展出一种新型高效合成多取代吡啶类衍生物的方法.该方法具有反应选择高、催化剂廉价无毒、条件温和、无污染等优点,为该类化合物的合成开辟了一条新途径.
