应用全外显子测序对有汗性外胚叶发育不良家系致病基因突变筛查
2018-11-28张淑娟梁景耀刘玉梅
张淑娟,梁景耀,田 歆,刘玉梅
(广州市皮肤病防治所 皮肤科, 广东 广州510095)
有汗性外胚叶发育不良综合征(Hidrotic ectodermal dysplasia,HED),又称Clouston syndrome(OMIM no.129500),是一种以甲营养不良、毛发缺陷和掌趾角化(或牙齿发育不良)等三联征为特征的遗传性综合征,属常染色体显性遗传[1]。
既往研究认为,HED的致病基因定位于13q11-q12.1,为编码人缝隙连接蛋白Cx30(connexin30)的GJB6(gap junction protein beta 6)[2]。目前发现患者GJB6有四个突变位点与疾病明确相关,包括G11R,V37E,D50N和A88V[3]。
近期笔者收集了一个HED的家系,家系患病成员具有典型的三联征表型,包括甲营养不良、毛发缺陷和掌趾角化,但经全外显子测序及Sanger测序的方法并未发现GBJ6突变,而发现有GBJ2基因的突变异常。
1 资料与方法
1.1 临床资料
家系情况见图1,家系中患儿(III1)为典型的HED患者,其母(II2)及舅舅(II3)表型严重程度不一,外显不全。其父(II1)、爷爷(I2)、奶奶(I1)、外公(I4)、外婆(I3)为正常表型。
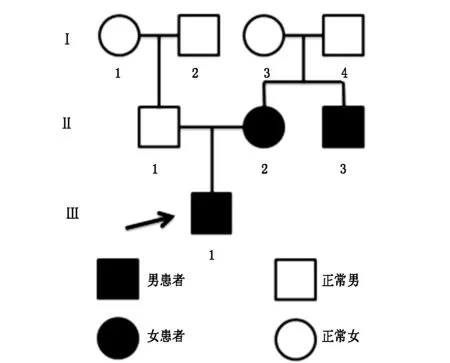
图1 HED综合症患者家系图(图中箭头所示为先证者)
患儿(III1),3岁,患手掌足底红斑角化3年。患儿出生时无头发、眉毛生长,全身毳毛稀疏。半岁时足底出现指甲盖大小红斑,逐渐扩大,并在红斑上出现角化,红斑角化进行性加重,渐累及双手手指,伴皲裂疼痛。趾甲自1岁起出现甲营养不良改变。体格检查:生长发育迟缓,身高85 cm,体重12 kg。语言障碍。视力、听力正常,牙齿排列整齐。系统检查无异常。皮肤科检查:全身皮肤毳毛稀少,头发缺失,头皮光滑,眉毛、睫毛均缺如。双手手指可见境界清楚的红斑角化,覆鳞屑,部分剥脱,伴皲裂。双足前端见红斑角化,覆厚层黄色角化性斑块,伴皲裂,左足为甚。部分趾甲灰黄,上见凹痕。
其母(II2)自幼无头发、眉毛生长,伴趾甲灰黄、粗糙,凹凸不平,但掌趾角化不明显。其舅(II3)自幼无头发生长,指甲正常,无掌趾角化。
1.2 方法
1.2.1全外显子组测序
经过知情同意后,取患儿(III1)外周血4 ml,EDTA抗凝,负80℃保存。采用DNA提取试剂盒抽提基因组。全外显子组测序服务由华大基因提供。简要实验及分析流程:将基因组DNA打断后制备文库。外显子捕获下来,经PCR线性扩增后进行文库质检,合格即可进行测序。根据库检合格文库的有效浓度及数据产出需求进行Illumina HiSeq 4000 PE150测序。获得原始测序序列后,去除低质量的数据以及人群中高频率的变异位点。
1.2.2Sanger测序验证
经过知情同意后,取患儿(III1)、患儿母亲(II2)、舅舅(II3)、外婆(I3)以及外公(I4)外周血4ml,EDTA抗凝,负80℃保存。采用DNA提取试剂盒抽提基因组。Sanger测序利用双脱氧末端终止法的原理,测序服用由华大基因提供。测序位点为GBJ6的四个经典突变位点,G11R(rs104894415),V37E(rs104894416),D50N(CM085977),A88V(rs28937872)。以及检测GBJ2的突变位点235delC(rs80338943)。
2 结果
2.1 患儿全外显子组测序结果
对患儿进行全外显子组测序,发现编码缝隙连接蛋白Cx26的GBJ2基因发生突变[4],突变位点为rs80338943,又名235delC。
HED经典的致病基因GBJ6的四个位点G11R(rs104894415),V37E(rs104894416),D50N(CM085977),A88V(rs28937872)均无突变。另外,既往报道的GJA1基因的V41L位点,以及GJB2的R127H、F191L位点未发现突变[5,6]。
2.2 家系Sanger测序结果
患儿(III1)、患儿母亲(II2)、舅舅(II3)、外婆(I3)以及外公(I4)5个样本经Sanger测序验证,得出以下结果。
在5个样本中,GBJ6的4个经典突变位点,G11R(rs104894415),V37E(rs104894416),D50N(CM085977),A88V(rs28937872)均未发生突变。
GBJ2在患儿(III1)、患儿母亲(II2)、外婆(I3)中发生突变,突变位点均为rs80338943。患儿突变序列为GCTATGGGCC[-/C]TGCAGCTGAT。患儿母亲突变序列为GCTATGGGCC[C/-]TGCAGCTGAT,外婆突变序列为GCTATGGGCC[C/-]TGCAGCTGAT。外公(I4)以及舅舅(II3)在该位点未发现突变(图2)。
3 讨论
外胚叶发育不良(ectodermal dysplasias,ED)是一组临床表型多样的遗传性疾病,主要累及头发、牙齿、指甲和皮肤[7]。其中HED是最为常见的类型,并以甲营养不良、毛发缺陷和掌趾角化三联征为特征。受遗传和环境的影响,患者的表型可有异质性,有时仅有其中1-2种临床表现[8]。在本研究中,患儿具有典型的三联征表型,可以明确诊断为HED。而母亲只有普秃和指甲损害,掌趾角化不明显;其舅只有普秃。该家系的3名患者符合HED表型多样的特点。
缝隙连接蛋白(connexin,Cx)是构成细胞间缝
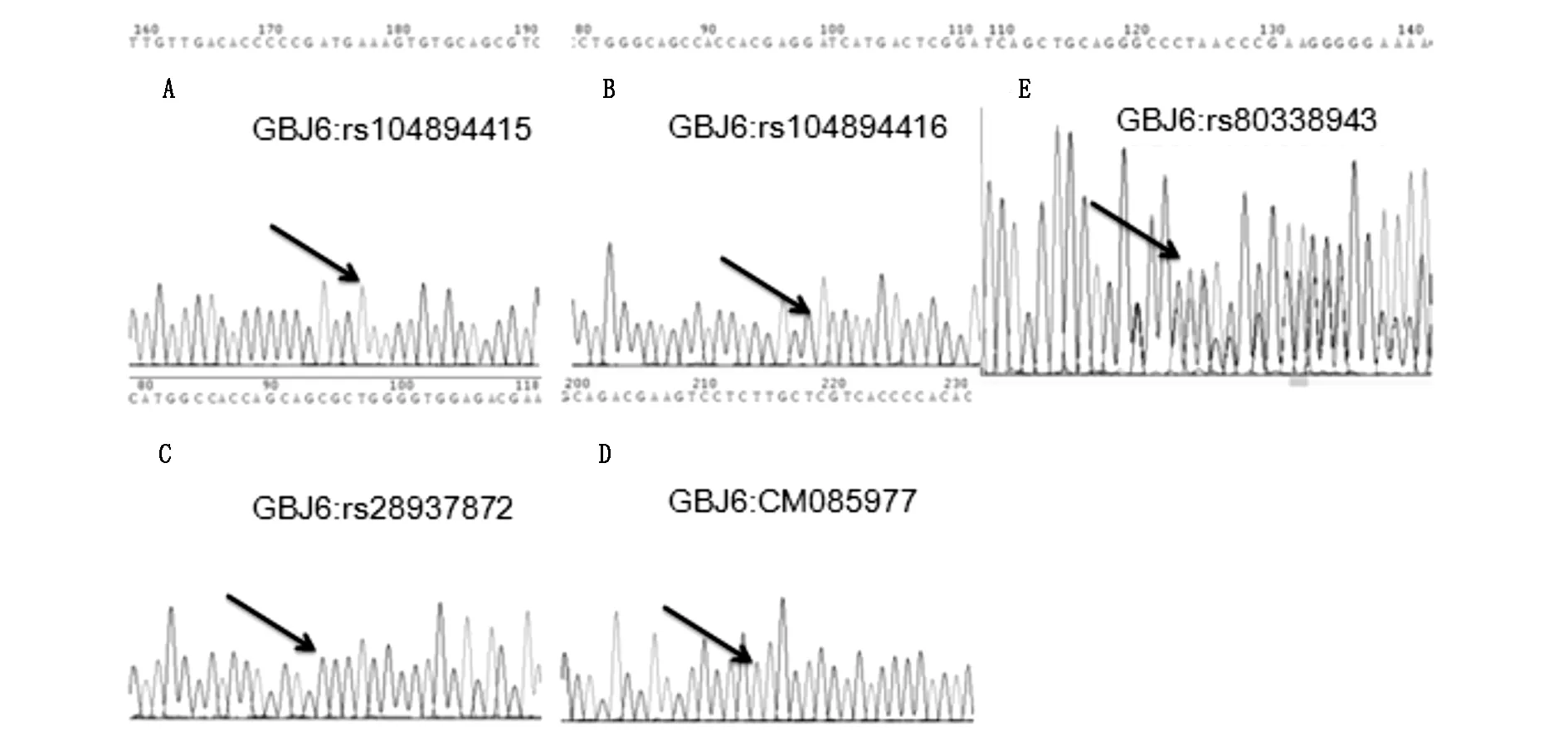
图2 Sanger测序结果。A-D为患儿(III1)GBJ6四个位点的检测结果;E显示患儿(III1)GBJ2的rs80338942位点发生突变
隙连接(gapjunction,GJ)通道的基本结构和功能的一大类膜蛋白。它们参与细胞间信号的传导,并在维持皮肤平衡中发挥关键的作用[9]。既往大部分研究中,编码Cx30的GJB6被认为是HED的重要致病基因之一,其四个位点的突变(G11R,V37E,D50N和A88V)与疾病明确相关[1-3]。而在本研究中,虽然患儿(III1)有典型的HED三联症的表型,但无论采取全外显子测序还是Sanger测序,并未发现GJB6(位点G11R,V37E,D50N和A88V)的突变,其余两例不完全表型的患者(II2,II3)亦无GJB6突变。该结果提示GJB6并不是HED必需的或者唯一的致病基因。
外显子组的序列仅占全基因组序列的1%左右,但85%致病突变位于外显子区。为了探索本HED家系中是否存在GJB6以外的致病突变,本课题组进行了全外显子测序,即利用序列捕获技术将全基因组外显子区域DNA捕捉并富集后进行高通量检测[10],结果发现GBJ2在患儿中发生突变。并进一步用Sanger测序的方法确认患儿(III1)、患儿母亲(II2)、外婆(I3)均携带GBJ2突变,突变位点均为rs80338943(又名235delC)。GBJ2编码Cx26,与GJB6类似,也是编码缝隙连接蛋白。GJB2主要表达于皮肤和内耳,其突变可引起皮肤异常及耳聋[9]。在过往的研究中,发现GJB2的R127H、F191L位点发生突变,与HED相关[5,6]。而GJB2基因235delC位点突变与HED的关系暂未见报道。然而,家系中舅舅(II3)未携带GJB2突变基因而有普秃表型,而外婆表型正常而携带GJB2基因突变,提示GJB2基因235delC位点的突变与HED的关系有待进一步确认。另外,有报道发现,GJA1基因(编码Cx43)V41L位点的突变与HED的发病相关[6],但在本研究的全外显子测序结果中亦没有发现GJA1的突变。
本课题报道了一个具有典型HED表型的家系,并通过全外显子测序及Sanger测序,筛查HED家系的致病基因,发现HED患者并没有携带既往报道的GJB6(G11R,V37E,D50N和A88V)、GJB2(R127H、F191L)以及GJA1(V41L)基因位点的突变。本研究为探明HED疾病的相关基因及分子机制提供新的线索和思路。
