PNPLA2基因新发纯合移码突变导致中性脂肪沉积症伴肌病1例报告
2018-11-14卢晓庆胡波琳陈燕红丁卫江漆学良
卢晓庆, 胡波琳, 陈燕红, 丁卫江, 漆学良
中性脂肪沉积症(neutral lipid storage disease,NLSD)为一种常染色体隐性遗传的脂质沉积性肌病,包括中性脂肪沉积症伴鱼鳞病(neutral lipid storage disease with iehthyosis,NLSDI)和中性脂肪沉积症伴肌病(neutral lipid storage disease with myopathy,NLSDM)。前者在我国尚未见报道。后者是由PNPLA2(patatin-like phospholipase domain-containing protein 2,PNPLA2)基因突变致脂肪甘油三酯水解酶(adipose tridyceride lipase,ATGL)功能缺陷而导致的代谢性肌病,该病于2007年由法国的Fischer等[1]首次报道。截至2016年,国内外累计报道经基因检测证实的NLSDM病例不超过50例,其中中国患者仅数例[2]。目前中国报道的该病患者多来自中国北方和西南地区,我们报道1例中部地区PNPLA2新发纯合移码突变导致的NLSDM。
1 临床资料
患者,女性,33岁,因“右下肢无力2 y余,加重伴右上肢及左侧肢体无力6 m”入院。患者2 y前无明显诱因逐渐出现右下肢无力,表现为上楼抬腿费力,平地行走1500米左右,即需休息数分钟方能继续行走,未重视,未处理。近6 m来,患者右下肢无力较前加重,上楼需搀扶,平地行走数百米即需休息,同时逐渐出现右上肢及左侧肢体无力,上肢抬举费力、穿脱衣困难,右上肢明显(见图1)。上诉症状无晨轻暮重现象,无肢体麻木疼痛、肉跳;无睑下垂、吞咽困难、构音障碍;无发热、心悸、胸闷、皮疹、关节痛。自起病来,患者一般情况好,二便未见异常。既往体健,否认家族史。体格检查:一般内科查体无明显异常。神经系统查体:神志清楚,言语流利,高级智能活动正常,脑神经检查无明显异常,感觉检查正常。抬头力弱,颈伸肌肌力Ⅳ-级,颈屈肌肌力Ⅳ+级,右三角肌肌力Ⅱ级,左三角肌肌力Ⅳ+级,右肱二头肌肌力Ⅲ-级,左肱二头肌肌力Ⅳ级,双上肢远端肌力Ⅴ级,双侧髂腰肌Ⅲ级,双下肢近端肌力Ⅳ级,远端肌力Ⅴ级,右侧三角肌、冈上肌、冈下肌萎缩,四肢肌肉无肥大,无肌肉压痛,未见肌束颤动。共济运动正常。双侧肱二头肌反射、膝反射(+)。双侧病理征阴性。脑膜刺激征阴性。辅助检查:肌酸激酶1754.93(正常值40~200)U/L,肌酸激酶同工酶78.46(正常值0~24)U/L,肌红蛋白610.14(正常值0~100)μg/L。血清总胆固醇5.3(正常值3.0~5.2)mmol/L。外周血涂片油红“O”(ORO)染色示:在多核细胞的胞质内可见较多脂肪滴沉积(见图2)。肌电图检查示:双侧肱二头肌肌源性损害。肌肉病理检查(右肱二头肌):HE染色示肌纤维大小不均,大量肌纤维内可见大小不一的筛状空泡形成,可见少许坏死、再生肌纤维,坏死肌纤维内见炎细胞浸润;ORO染色显示空泡肌纤维内脂肪滴大量聚集,少数融合成片;在MGT染色下可见含有空泡的肌纤维略红染,可见较多镶边空泡(rimmed vacuoles,RV)(见图3)。基因测序分析:c.750_757del(p.Q250fs)纯合移码突变(见图4)。

图1 NLSDM患者双上肢抬举费力,两侧不对称
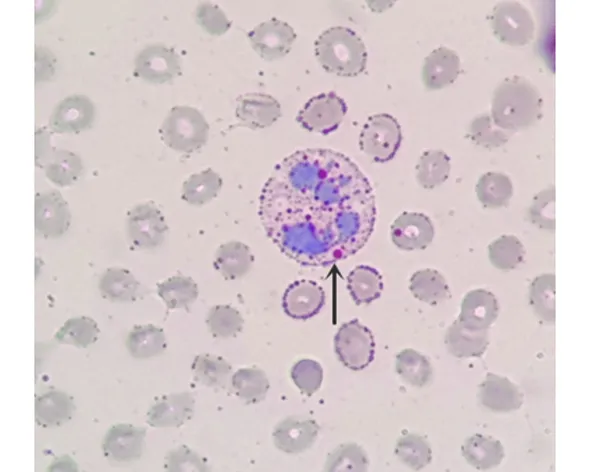
图2 NLSDM患者外周血涂片(ORO染色×1000)。可见粒细胞的胞质内脂肪滴沉积(Jordan小体)
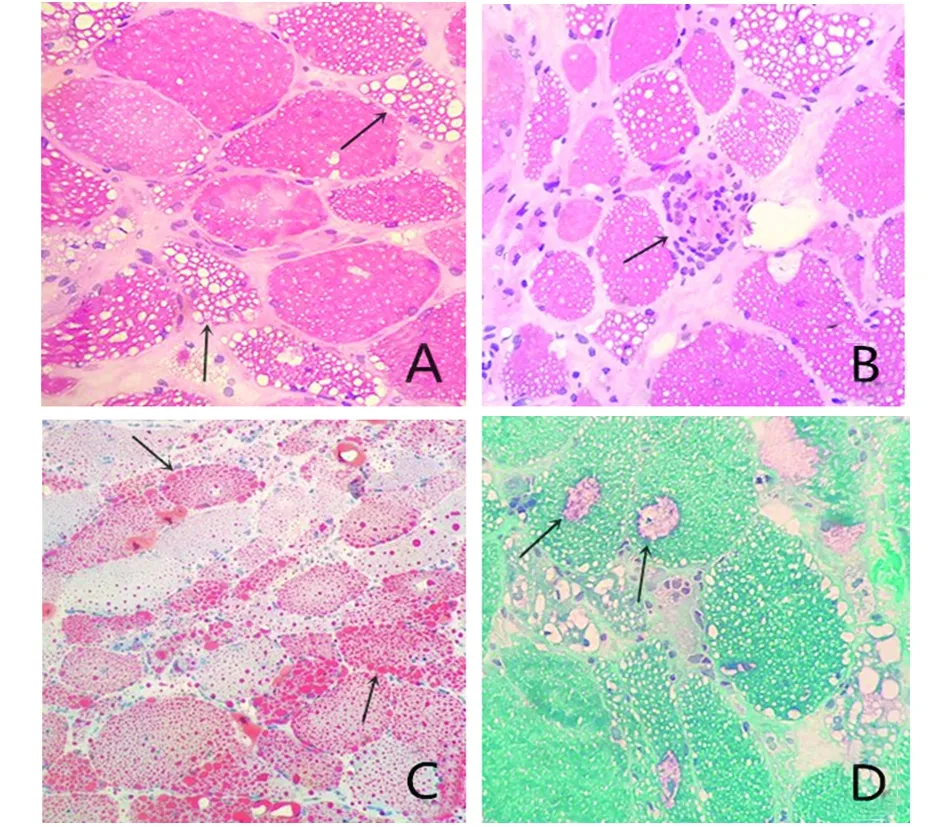
图3 A:肌纤维内大量脂质空泡,大小不等(HE×400); B:坏死伴吞噬肌纤维(HE×400); C:肌纤维内大量脂滴沉积(ORO×400); D:肌细胞可见较多镶边空泡(改良Gomori三色法×400)
图4 c.750_757del(p.Q250fs)纯合移码突变。参考序列 NM_020376
2 讨 论
NLSDM患者多在成年后起病,平均年龄30岁,起病隐匿,缓慢进展[3]。其临床异质性较大,可出现无症状高肌酸激酶血症[4]、运动不耐受、肌无力,甚至有些患者会因心脏损害[5]而死亡。患者可有近端和远端肢体无力,近端重于远端,上肢重于下肢。早期肌无力现象可不对称,右上肢无力更为突出,可能与多数患者的右利手有关,但是随着疾病的进展,双侧无力基本对称[2]。本例患者起病早期表现为不对称性肢体无力,右上肢无力明显,这是NLSDM的特点之一。NLSDM患者心脏受累也较多见,部分患者还可伴有高脂血症、肝肿大、糖尿病等,极少数可累及神经系统而出现认知功能障碍[6]。
实验室检查外周血涂片ORO染色对该病诊断具有较高的价值,几乎在所有患者的中性粒细胞胞质内可见脂肪滴沉积,也称为Jordan小体[7]。血清肌酸激酶多轻至中度升高。肌电图检查呈肌源性改变。影像学肌肉磁共振检查可见脂肪浸润肌肉组织,下肢受累比上肢重,且其不对称性更大[8]。肌肉组织活检典型的病理改变是肌纤维大小不一,骨骼肌纤维内可见大量的脂滴沉积,坏死、再生肌纤维及炎细胞浸润少见。部分患者肌纤维内可见镶边空泡。镶边空泡特异性相对较差,还可见于其他多种肌病[9]。但它的出现有助于与其他类型的脂质沉积性肌病相鉴别。
基因检查可明确NLSDM的诊断,PNPLA2基因为该病的致病基因,该基因编码的蛋白质为脂肪甘油三酯水解酶,即PNPLA2蛋白,该蛋白在人体组织中广泛表达,致病性突变导致中性脂肪在外周血粒细胞、骨髓、骨骼肌、心肌及肝脏中异常沉积[10]。PNPLA2蛋白的N’端是甘油三酯水解位点,由1~5号外显子编码,该结构域基因突变往往引起严重的肌病伴其他系统损害;C’端是脂肪结合位点,由6~10号外显子编码,该结构域基因突变仅导致骨骼肌损害,PNPLA2基因突变主要集中在第4~7号外显子[11,12]。PNPLA2基因可分为纯合突变和复合杂合突变,突变类型有缺失、错义、无义和重复突变,其中缺失突变占大多数[13]。最近Tan等[2]报道的4例中国西南地区NLSDM患者均为纯合突变。
我们报道的该例患者突变位点定位于11号常染色体PNPLA2基因的6号外显子,即C’端结构域,携带c.750_757del(p.Q250fs)纯合移码突变,可见cDNA第750_757碱基缺失引起氨基酸第250位谷氨酰胺之后编码序列改变,该突变导致编码的蛋白质PNPLA2紊乱,丧失正常功能。患者临床仅出现骨骼肌损害,符合NLSDM的临床表型。PLPNA2基因为常染色体隐性遗传,理论上必须在两个等位基因上均发生突变才有可能致病(纯合或复合杂合突变),该患者在此基因外显子区域上发现了一个纯合突变点,理论上可能致病,而且移码突变导致编码的蛋白质PNPLA2紊乱,丧失正常功能,是致病性的极强证据,因此我们推测其为致病性突变。查阅人类基因突变数据库(HGMD数据库)及相关文献,发现该突变位点尚未见报道。我们为首次报道该新发突变。
相比于其他类型的脂质沉积性肌病,目前该病尚无特殊治疗。激素、核黄素、辅酶Q10、左旋肉碱治疗效果均不明显。患者应避免饥饿、寒冷和长时间运动。推荐高碳水化合物饮食以保持足够的糖原储存。中链脂肪酸饮食疗法可能有一定疗效,其在椰子油、棕榈仁油、黄油、牛奶、酸奶及奶酪中占比较高[14],可适当增加此类食物的摄入。
总之,NLSDM发病率低,临床罕见。主要累及骨骼肌和心肌,多为成年起病,缓慢进展,肌肉受累程度不对称,近端和远端均可累及,外周血涂片ORO染色可见Jordan小体,肌肉活检和基因检查可确诊。目前,该病尚无有效治疗手段,预后通常良好。
