抑制PKCβ2活化改善脂多糖诱导的大鼠肺微血管内皮细胞线粒体损伤
2018-11-09夏中元苏娃婷雷少青
张 元,夏中元,苏娃婷,雷少青
(武汉大学人民医院麻醉科,武汉 430060;*通讯作者,E-mail:xiazhongyuan2005@aliyun.com)
脓毒症被定义为由于宿主对感染的反应失调而危及生命的器官功能障碍[1]。其中,急性肺损伤(acute lung injury,ALI)/急性呼吸窘迫综合征(acute respiratory distress syndrome,ARDS)是脓毒症高死亡率的突出原因[2]。然而,积极地抗炎治疗并未取得预期的效果。鉴于越来越多的证据显示ALI/ARDS的一个显著特征是肺微血管内皮屏障完整性受损,恢复内皮屏障完整性可能是有效的治疗方法[2]。既往研究表明,线粒体来源的活性氧簇(mitochondrial-derived reactive oxygen species,mtROS)通过协同促进内皮细胞表达促炎症因子,可能是内皮细胞功能障碍的重要机制[3,4]。新近研究发现,PKCβ2是调控mtROS生成的关键上游信号[5,6]。据此,本研究拟探讨PKCβ2信号在LPS诱导的RPMECs线粒体损伤中的作用机制。
1 材料与方法
1.1 主要材料和试剂
RPMECs购自武汉大学细胞典藏中心。LPS购自美国Simga公司。磷酸盐缓冲液(phosphate buffered saline,PBS)、DMEM低糖培养基、胎牛血清购自美国Gibco公司。青霉素-链酶素溶液(双抗)、胰酶消化液购自生物科技公司(BIOSHARP)。LDH活性检测试盒(酶标仪法)、Cell Counting Kit-8试剂盒、SOD和MDA检测试剂盒购自南京建成生物工程研究所。Western抗体稀释液、蛋白marker购自碧云天生物技术研究所。p-PKCβ2、PKCβ2、Cyt c、GAPDH一抗及二抗购自美国Cell Signaling公司。
1.2 细胞培养与药物干预
RPMECs培养于含10%胎牛血清和1%双抗的DMEM低糖培养基;置于5% CO2、37 ℃细胞培养箱,隔天换液。生长良好的细胞随机分为三组(n=6):正常对照组(C组),脂多糖处理组(LPS处理组),脂多糖处理加PKCβ2特异性抑制剂CGP53353治疗组(LPS+CGP组)。细胞贴壁生长24 h后,无血清培养基同步化2 h,随后更换含1%胎牛血清和1%抗生素的DMEM低糖培养基,并加入同时药物干预24 h。LPS终浓度为10 μg/ml;CGP终浓度为1 μmol/L。
1.3 LDH含量检测
收集6孔板中培养基于1.5 ml EP管中,14 000×g4 ℃离心10 min,取上清液,按LDH检测试剂盒说明书操作,酶标仪检测各组吸光度值,波长设定为450 nm。
1.4 细胞活力检测
96孔板中的细胞,预冷PBS洗涤3次后,每孔加入无血清DMEM低糖培养基稀释的CCK-8工作液100 μl,37 ℃孵育2 h,酶标仪检测各孔吸光度值,波长设定为450 nm。
1.5 SOD活性及MDA含量检测
6孔板中细胞,预冷PBS洗涤3次;加入RIPA强效裂解液,4 ℃静置30 min后,14 000×g4 ℃离心10 min,收集上清液,BCA法测定细胞总蛋白浓度。相应商品化试剂盒检测细胞总蛋白中SOD活性及MDA含量。
1.6 Western blotting分析
调整细胞总蛋白样品浓度后,按每孔50 μg上样,聚丙烯酰胺凝胶电泳分离蛋白后,湿法转移至PVDF膜。PBS洗涤3次,每次10 min,用5%脱脂奶粉室温封闭2 h。TBST再次充分洗涤后,按marker剪裁出目标蛋白所在区域条带,置于相应一抗(p-PKCβ2、PKCβ2、Cyt c、GAPDH)4 ℃低速摇床孵育过夜。取出后TBST再次充分洗涤,置于相应二抗室温避光孵育2 h。随后再次洗涤,置入Odyssey远红外荧光扫描成像系统(美国,LI-COR公司)扫描分析。
1.7 统计学方法
采用GraphPad Prism 6.01软件进行数据分析,所有数据以均数±标准差表示,组间比较采用单因素方差分析(one-way ANOVA)。P<0.05为差异具有统计学意义。
2 结果
2.1 CGP对LPS环境下大鼠肺微血管内皮细胞活力和LDH释放量的影响
与正常对照组比较,LPS暴露可导致RPMECs细胞活力显著降低(P<0.05),且培养基中LDH含量显著增加(P<0.05)。CGP治疗后,细胞活力显著恢复(P<0.05),LDH释放量显著减少(P<0.05),但仍显著高于正常对照组(P<0.05,见图1)。
2.2 CGP对LPS环境下大鼠肺微血管内皮细胞SOD活性及MDA含量的影响
与正常对照组比较,LPS暴露可导致RPMECs内SOD活性显著降低(P<0.05),且MDA含量显著增加(P<0.05)。CGP治疗可有效逆转上述变化(P<0.05),但仍与正常对照组具有统计学差异(P<0.05,见图2)。
2.3 CGP对LPS环境下大鼠肺微血管内皮细胞PKCβ2活化和Cyt-c表达的影响
与正常对照组比较,LPS暴露可导致RPMECs内PKCβ2显著活化,表现为p-PKCβ2比率显著上升(P<0.05);同时可导致Cyt c蛋白表达水平显著上调(P<0.05)。CGP治疗可有效逆转上述变化(P<0.05),但仍显著高于正常对照组(P<0.05,见图3)。
3 讨论
本研究通过构建RPMECs的LPS损伤模型,并应用PKCβ2特异性抑制剂干预,首次探讨了PKCβ2信号在LPS诱导的RPMECs线粒体损伤中的作用及其相关机制。
微血管内皮细胞间相互连接,形成半渗透性屏障结构,可动态调节血管内外物质交换过程。然而,革兰氏阴性细菌内毒素的主要毒性物质LPS可通过诱导内皮细胞变形和内皮细胞间隙形成,进而导致内皮细胞屏障结构破坏,通透性增加[7]。临床研究发现,肺微血管内皮屏障功能障碍是各种高死亡率疾病(如:脓毒症相关的ALI/ARDS)发生、发展的关键环节。然而,针对改善肺微血管内皮细胞屏障功能的具体药物治疗仍然缺乏。因此,深入探究肺微血管内皮细胞屏障调节机制,有利于优化脓毒症相关的ALI/ARDS的防治策略[7]。
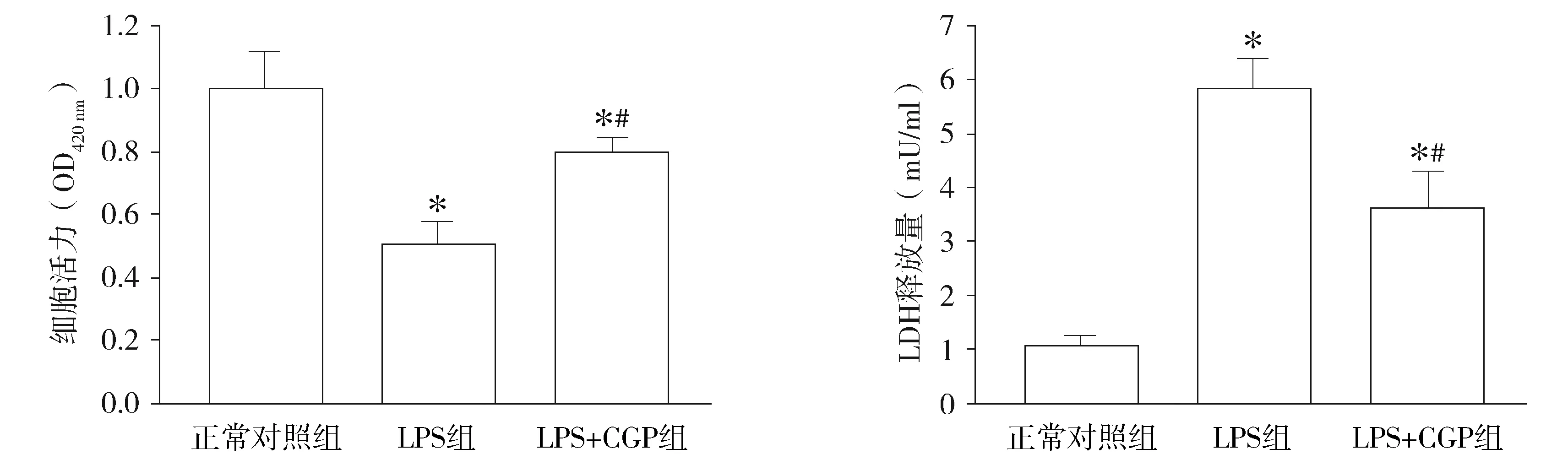
与正常对照组比较,*P<0.05;与LPS组比较,#P<0.05

与正常对照组比较,*P<0.05;与LPS组比较,#P<0.05

与正常对照组比较,*P<0.05;与LPS组比较,#P<0.05
蛋白激酶C(protein kinase C,PKC)是一类多功能的丝氨酸/苏氨酸激酶;PKCβ是其重要成员,包括β1和β2两个亚型。既往研究指出,PKC活化可导致微血管内皮细胞肌动蛋白聚合,并诱导内皮细胞连接解离,局部粘连结构重排,进而导致内皮细胞通透性增加[8]。进一步研究发现,抑制微血管内皮细胞PKCβ活化,可通过降低细胞内Ca2+水平,抑制内皮细胞皱缩,进而改善内皮细胞通透性[9]。而PKCβ2高度活化已被证明是糖尿病肾病及糖尿病视网膜病变血管高通透性的重要机制[10]。本研究亦发现,LPS可诱导RPMECs内PKCβ2高度活化;而抑制PKCβ2活化具有显著的内皮细胞保护作用。
新近研究表明,线粒体在炎症反应中具有重要作用。线粒体不仅是模式识别受体信号转导和调控的重要平台;而且,应激诱导的线粒体组分外渗是损伤相关分子模式免疫应答的重要初始环节[11]。其机制与mtROS介导的促炎症因子表达上调密切相关[12]。最新研究发现,LPS可导致人脐静脉内皮细胞线粒体膜电位降低、Cyt c外渗,线粒体过度分裂呈碎片化,进而诱导线粒体途径的细胞凋亡[13]。本研究结果显示,LPS暴露导致的RPMECs线粒体损伤,涉及氧化应激增强、Cyt c表达上调等机制。
有意义的是,新近研究发现,脑缺血再灌注后,PKCβ2活化并转位至线粒体,可能是海马缺血耐受区的重要保护机制[14]。虽然活化的PKCβ转位至线粒体后,对线粒体功能的影响尚有争议;但是,已有研究指出,PKCβ依赖的p66Shc磷酸化,是p66Shc线粒体转位,进而通过氧化还原型Cyt c、介导mtROS生成的关键环节[5];而且,抑制PKCβ活化,可减少p66Shc磷酸化,并抑制其转位至线粒体,进而减少mtROS生成,减轻细胞氧化损伤[6]。在本研究中,抑制PKCβ2活化,亦可显著改善LPS诱导的RPMECs线粒体损伤。
综上所述,本研究证实了抑制PKCβ2活化可改善LPS诱导的RPMECs线粒体损伤;但其机制是否与PKCβ2线粒体转位有关,仍有待进一步研究。
