Krüppel样转录因子2的功能
2018-10-29荆堂堂倪晋泽赵玉菲朱亮
荆堂堂, 倪晋泽, 赵玉菲, 朱亮
(1.大连医科大学 基础医学院 生理教研室, 辽宁 大连 116044; 2.安徽医科大学 第一附属医院 胸外科, 安徽 合肥 230022)
血管内皮健康状况对于血管的正常生理和功能关重要.血管内皮细胞是多种生理刺激的感受器和效应器,参与调节选择通透性,血液凝固以及免疫细胞归巢等多种生理过程.血管内皮功能失调导致的微循环障碍是各种组织以及器官缺血缺氧损伤的共同特征.而血流剪切力可诱导一些血流依赖性转录因子的表达来抵抗血管内皮细胞的损伤.特别是依赖血流产生的KLF2对维持血管内皮的功能至关重要[1].Krüppel样转录因子2(Krüppel-like transcription factor 2,KLF2) 作为KLF家族中重要的转录因子,在心血管领域, 特别是在内皮功能调控中所起作用,已成为研究热点.
1 KLF家族(KLFs)
KLFs是DNA结合转录因子家族的1个锌指亚类,其命名是基于DNA结合域与果蝇蛋白质Krüppel同源,“Krüppel”为德文“残疾”, 此蛋白质被命名是由于果蝇胚胎纯合子表达的Krüppe蛋白使其前腹、胸段发生改变,导致其死亡[2].第1个Krüppel因子(KLF1/EKLF)被发现于哺乳动物的红细胞,后来发现此因子在β珠蛋白基因的合成和红细胞的发育中起着重要的作用.从1993 年EKLF的最初发现,共发现17种哺乳动物的Krüppel,根据发现的时间顺序分别命名为KLF1-KLF17[3].
2 KLF2的结构特点
Lingrel等利用EKLF/KLF1锌指结构域的同源性,首次克隆出转录因子KLF2,KLF2是由354个氨基酸组成的蛋白,在肺组织中高表达,最初被称为肺KLF样因子(lung Krüppel-like factor,LKLF).人类KLF2定位于染色体的10p13.1,其与鼠基因的同源性大于85%.位点分析显示在KLF2的启动子区域内长约75 bp的高度保守序列,含有肌细胞增强因子2(myocyte enhancing factor 2,MEF2)的结合位点,是调节KLF2表达的关键位点[4-5].KLF2编码区有1655个碱基,编码354个氨基酸,其N端的第1~110位氨基酸为转录激活区,第110~267位氨基酸为转录抑制区(图1)[5].KLF2转录抑制区可以与泛素连接酶 WWP1(WW domain-containing E3 ubiquitin protein ligase 1)结合,致使KLF2泛素化和蛋白酶体降解[6].
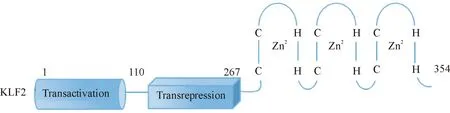
图1 KLF2结构示意图
3 KLF2的血流依赖性调节
在血流依赖性转录因子KLF2的转录起始点上游的-157~-79 bp区域含有一段高度保守序列,包含1个MEF2的结合位点[7]. 当存在正常的血流时,在血流产生的剪切力的刺激下,KLF2在转录水平激活,随后KLF2 mRNA和蛋白表达增加.血流产生的剪切力可促进KLF2的表达,不仅是在转录水平激活KLF2的启动子,也可以使KLF2的mRNA稳定[8].在转录水平激活KLF2启动子的关键是肌细胞增强因子2(myocyte enhancing factor 2,MEF2)结合到KLF2上的MEF2结合位点[9].MEF2的激活,一方面是通过上游丝裂原活化蛋白激酶(mitogen activated protein kinase,MAPK)信号的级联放大,包括MAPK激酶5(MAPK kinase 5,MEK5)和细胞外信号调节激酶5(extracellular-signal-regulated kinase 5,ERK5)[10];另一方面是通过解除组蛋白脱乙酰酶5/7(histone deacetylase 5/7,HDAC5/7)对KLF2转录活性的抑制,即当存在正常的血流时,血流剪切力可以通过激活钙离子/钙调蛋白依赖的蛋白激酶(Ca2+/calmodulin-dependent manner,CaMK)信号通路,通过MEK5/ERK5/MEF2通路,在转录水平激活KLF2,随后KLF2蛋白表达,使HDAC5/7磷酸化,改变了HDAC5/7的细胞定位,使其从细胞核输出到胞质,解除对MEF2的抑制,促进KLF2的表达,增加内皮细胞保护因子的表达(图2)[11].
4 KLF2对血管内皮的作用
KLF2在血流介导的多基因表达调控过程中发挥重要作用.在血流作用下基因的表达上调有15%是KLF2依赖性的[9].有研究表明KLF2连同核因子Nrf-2调控着约70%的剪切力诱导的内皮细胞基因的表达[10].KLF2在调节血管张力、止血/血栓形成、炎症、调节内皮屏障功能和抗氧化能力等基因的表达过程中发挥重要作用(图3).KLF2在动脉粥样硬化发生和维持血管稳态之间扮演分子开关的角色,可直接影响动脉粥样硬化的发病机制[9,12].在ApoE基因敲除时,KLF2+/-鼠出现饮食诱导性的动脉粥样硬化,与ApoE基因敲除稳定的动脉粥样斑块相比,不稳定斑块的KLF2表达水平明显降低[12].
4.1 血管张力
健康的血管内皮细胞可通过产生一氧化氮(nitric oxide,NO)、C型钠尿肽(C-natriuretic peptide,CNP)、肾上腺髓质素等血管舒张因子和内皮素-1(endothelin-1,ET-1)、血管紧张素转换酶(angiotensin-converting enzyme,ACE)等血管收缩因子来调节血管张力[13],KLF2可强效诱导内皮型一氧化氮合酶(eNOS)和CNP的表达,抑制ET-1和ACE的表达.然而, KLF2还可抑制肾上腺髓质素的表达[14],KLF2可直接结合到eNOS基因启动子增加其转录活性和下调窖蛋白-1(caveolin-1,eNOS的负性调节因子)[9,15].
eNOS和NO在内皮细胞有舒血管作用,还可通过抑制内皮黏附分子ICAM-1和VCAM-1发挥抗炎和抗血栓作用,血流通过KLF2诱导eNOS的产生,表明剪应力是介导内皮细胞功能的一个重要机制[16].
4.2 血栓
KLF2可调控血栓相关性因子的表达,具有抗血栓作用.血栓调节蛋白(thrombomodulin,TM)是凝血酶诱导抗凝蛋白-C激活的关键辅因子,KLF2可直接与TM的启动子结合增加TM的活性并促进其表达,KLF2过表达抑制促凝因子-纤溶酶原激活物抑制剂(plasminogen activator inhibitor,PAI-1)、血管性血友病因子(von Willebrand factor,vWF)和组织因子(tissue factor,TF)的表达,轻度增加组织型纤溶酶原激活物(tissue plasminogen activator,tPA) 的表达;体外实验表明,人脐静脉内皮细胞KLF2过表达可明显增加凝血时间,相反,当KLF2敲除具有抗血栓作用的基因表达减少,促凝因子生成增加[17].
相比另一组慢病毒介导KLF2过表达后,发现vWF 的mRNA和蛋白水平约呈两倍增加.凝血酶和毛喉素(vWF的诱导剂)刺激后,也可检测到vWF蛋白水平呈两倍增加[18].此外,Weibel Palade小体-血管内皮细胞vWF及其他生物活性化合物的贮存器过表达KLF2时,炎症因子血管生成素-2和白细胞介素-8(IL-8)表达减少,但与对照组相比,人脐静脉内皮细胞平均分布更多[19].
4.3 炎症
腺病毒介导的KLF2过表达可抑制白细胞介素-1β(IL-1β)、肿瘤坏死因子α(TNFα)、脂多糖(lipopolysaccharide,LPS)所介导的细胞黏附分子VCAM-1和E-选择素的表达,使T细胞的黏附和滚动作用减弱[9].KLF2不仅通过抑制炎性因子IL-1β的产生发挥抗炎作用,而且可通过与抑制核转录因子-κB(NF-κB)通路的多个组件相互作用,降低NF-κB的活性,间接发挥抗炎作用[9,17].凝血酶可与其受体结合,通过NF-κB通路介导炎症反应, KLF2可抑制凝血酶受体(protease-activated receptor-1,PAR-1)的表达,从而抑制凝血酶介导的NF-κB核积累与DNA的结合,降低炎症反应[20].
TNF-α通过NF-κB激活炎症信号,KLF2表达降低导致抗炎因子BMPER表达水平降低.低水平的BMPER使促炎因子炎性骨形态发生蛋白2(bone morphogenic protein 2,BMP2)表达增加,从而降低eNOS和增加ICAM-1和VCAM-1的表达水平,诱导白细胞黏附和外渗.在人血管内皮细胞KLF2的过表达,抑制TNF-α介导的BMPER因子的表达下调,使BMPER的表达增加,为KLF2在内皮细胞发挥的抗炎作用提供另一证据[21].
长期剪切应力介导的抗炎作用与抑制MAPK通路有关.JNK是AP-1激活组件c-Jun和ATF2的上游激酶.AP-1和NF-κB连同其他共激活物如CBP/p300形成转录复合物,可诱导血管内皮细胞炎性因子的表达,具有促炎、致动脉粥样硬化作用[22].KLF2的抗炎作用是通过肌动蛋白细胞骨架的变化来调节的,KLF2抑制参与细胞骨架调节JNK的磷酸化.结果显示JNK不能通过磷酸化被激活,同时也不能激活c-Jun和AP-1,MAPK通路被抑制,KLF2间接发挥抗炎作用[23].
4.4 补体激活
内皮细胞暴露于层流剪切应力时,内皮细胞表面的MAC抑制蛋白CD59(MIRL)的表达上调,使补体介导内皮细胞的溶解减少,剪切力作用是由ERK5/ KLF2信号通路介导的[24].
4.5 内皮屏障功能
KLF2+/-小鼠在炎症刺激下内皮渗漏增加[17].体外实验中,针对凝血酶,过氧化氢和组胺诱导的内皮细胞渗漏,KLF2过表达可通过上调紧密连接蛋白occludin的表达和降低肌球蛋白轻链的磷酸化,发挥保护内皮细胞的作用[25].在小鼠脑缺血模型中已经证实KLF2对内皮屏障功能的保护作用.为了研究小鼠大脑中动脉堵塞状况,对KLF2-/-小鼠,KLF2过表达小鼠和正常对照组小鼠大脑中动脉的梗死面积和血脑屏障功能进行分析,结果表明,由于occludin表达下降,KLF2-/-小鼠表现出较大的梗死面积和血脑屏障功能受损,KLF2过表达小鼠梗死面积减小和维持血脑屏障功能[26].
最近的研究表明,KLF2低表达可能参与阿尔茨海默病(Alzheimer’s disease,AD)病理过程[27].血脑屏障功能障碍和内皮通透性受损均与AD某段发病机理有关.在患AD的Tg 2576小鼠模型中,由于β淀粉样蛋白的累积,KLF2 的mRNA和蛋白水平表达明显减少,其机制为β淀粉样蛋白通过增加p53的表达水平,抑制KLF2的表达.人脑EC中 KLF2的高表达完全弥补了occludin表达的下降,与KLF2在内皮细胞屏障功能的作用一致.然而,在人体随机对照试验中,他汀类药物诱导KLF2的高表达,未能降低AD的发病率[28-29].
4.6 内皮细胞形态与细胞连接
血流和剪切力对血管内皮细胞骨架重排和细胞形态的影响已有约20的历史[30].KLF2在细胞骨架重排的重要作用已经被阐明.HUVECs 置于其排列方向与流动方向一致的剪应力(19±17 dyne/cm2,4 d)中.siRNA介导的KLF2沉默后,其排列方式改变,然而,即使在不存在血流的情况下,人脐静脉内皮细胞过表达KLF2后,由于伴随应力纤维形成的细胞骨架重排,其呈延伸状,而且细胞密度增加和病理迁移减少[18].
在高层流剪切应力条件下培养的内皮细胞连接蛋白37(CX37)高表达,其在ApoE基因缺陷小鼠会加速动脉粥样硬化过程[31].CX37是一种跨膜蛋白,在相邻细胞间以两个六聚体形式形成缝隙连接.缝隙连接是一种连接两个细胞胞质通过离子和小分子的物质交换来实现细胞间交流的特殊细胞连接.针对剪切应力或辛伐他汀诱导的KLF2过表达,其通过直接结合Cx37的启动子来调控Cx37的表达.KLF2敲除时,通过内皮细胞间缝隙连接的小分子和离子减少,表明KLF2在细胞间交流发挥的作用[32].
4.7 氧化应激
人脐静脉内皮细胞过表达KLF2后,使NAD(P)H、醌脱氢酶1(NAD(P)H dehydrogenase quinone 1,NQO1)、HO-1、GCLM及过氧化氢酶(catalase,CAT)等抗氧化基因表达上调,均为Nrf 2靶基因,其中一个主要的抗氧化转录因子是通过抗动脉粥样硬化血流被上调[18,33-34].KLF2可促进Nrf 2的核定位并激活其功能,增强Nrf 2介导的抗氧化应激作用[10].
HO-1是血红素分解代谢的限速酶,将其分解为胆绿素、一氧化碳和游离铁.此通路的产物在脉管系统具有抗氧化,抗炎抗凋亡作用[35].他汀类药物以KLF2依赖性方式强诱导HO-1的表达,通过HO-1激活后胆绿素和铁蛋白的产生抗氧化作用[36].
4.8 microRNA的产生
KLF2通过miRs在血管稳态中发挥重要作用.KLF2高表达时,调节细胞迁移的miR-150和miR-23b表达水平增加;而促进炎症反应的miR-155,miR-146a,miR-181a(决定内皮细胞命运)和miR-210(血管生成和缺氧反应)的表达水平减少.在剪应力和慢病毒介导的KLF2过表达时,具有抗动脉粥样硬化作用的miR-143/145表达上调.剪切力和/或KLF2可增加胞外囊泡的产生,使miR-143/145高表达,发挥抗动脉粥样硬化作用[37].
5 KLF2对非血管内皮的功能
5.1 T细胞和B细胞生物学
针对缺失位点分析显示,KLF2在T细胞静止和维持T细胞表型起着重要的作用[5].KLF2在胸腺细胞和T细胞的运输过程中发挥关键作用[6].T和B淋巴细胞在动脉粥样硬化过程中起着不同的作用[38-39].KLF2可诱导静止的单阳性T细胞(CD4+或CD8+)活化,但T细胞活化后KLF2迅速下降.KLF2缺乏小鼠外周单阳性T细胞大量减少,出现自发激活的细胞表型和Fas介导的细胞凋亡增加.KLF2可通过对原癌基因c-myc的负调控来维持T细胞静止.由于在ApoE基因缺陷小鼠CD8+T细胞的活化促进动脉粥样硬化易损斑块形成,KLF2维持T细胞表型具有生理重要性[40].此外,KLF2是外周T细胞再循环的必要因子.KLF2缺乏SP T细胞出现胸腺迁移受损和T细胞转运障碍;KLF2-/-T细胞大多数存在脾脏,血液或淋巴结内罕见,并证明KLF2通过诱导CD62L,β-整合素,CCR7(T细胞转运)和S1P1(胸腺细胞迁移)等关键受体的表达调节胸腺细胞和T细胞进入外周淋巴器官[41].
已知T细胞中的 CD4+CD25+fox3p+调节性T细胞(Tregs)有免疫耐受作用,对动脉粥样硬化的发生和发展有抑制作用.在OX-LDL作用下,Tregs以细胞接触的方式作用于血管内皮细胞,上调KLF2的表达[42].
在B细胞KLF2敲除导致转运分子CD62L和β-整合素表达降低,但S1P1受体表达几乎不受影响.KLF2敲除导致伴随边缘区B细胞亚群分化受损,动脉粥样硬化性B1细胞减少[43].
辛伐他汀在体外和体内可显著增加小鼠和人类T细胞KLF2的表达,导致IFN-γ的分泌和T细胞增殖减少,表明了KLF2在淋巴细胞的抗动脉粥样硬化作用[44].
5.2 单核细胞和巨噬细胞生物学
KLF2高表达于人类单核细胞,但当单核细胞在细胞因子或LPS作用下被激活分化成巨噬细胞时KLF2的表达降低.与对照组相比,动脉粥样硬化患者单核细胞中的KLF2表达减少约30%.因为单核细胞活化和募集在动脉粥样硬化中起重要作用. KLF2通过降低环氧合酶2(COX-2)、IL-1、IL-8、TNF-α和MCP-1的分泌,抑制LPS介导的单核细胞的激活.KLF2过表达也可降低单核细胞的吞噬活性,并不抑制,而是促进单核细胞向炎症部位的聚集.相反,KLF2敲除可增加单核细胞MCP-1,TF,和COX-2的表达,KLF2可通过与共激活因子PCAF相互作用,抑制NF-kB和AP-1转录活性,不改变NF-κB和AP-1的表达,核积累,或DNA结合,在单核细胞中发挥其抗炎作用[45].
巨噬细胞亚群:M1巨噬细胞(由GM-CSF、TNF或LPS激活)具有促炎作用,而M2巨噬细胞通常是抗炎的,有助于组织修复,但由于清道夫受体数量较多,积聚OX-LDL的能力更高[46].M2比M1型巨噬细胞的KLF2表达高.然而,接触OX-LDL后,M2型巨噬细胞KLF2表达降低,M1型巨噬细胞不变.在M2型巨噬细胞KLF2敲除导致MCP-1分泌增加[47]. 因之,KLF2在单核细胞和巨噬细胞生物学具有抗炎和抗动脉粥样硬化的作用.
5.3 血管生成
血管内皮生长因子(vascular endothelial growth factor,VEGF-A)是血管形成和血管新生的关键调节因子,其主要通过酪氨酸激酶受体VEGFR2发挥作用[48]. KLF2通过诱导内皮特异性miRNAs(miR-126)的表达在血管新生中起调节作用.具体机制为:miR-126可抑制VEGF通路的抑制剂Spred-1的活性,KLF2通过诱导miR-126的表达来间接促进VEGF-A/VEGFR2介导的血管新生[49].
血管生成素1(Ang-1)及其受体-酪氨酸激酶受体Tie2参与维持血管的静止和血管新生[50],机制为: 在细胞-细胞相互接触时,Ang-1诱导Tie2转录活性,优先激活Akt信号通路使血管静止.同时,Ang-1诱导Tie2锚定于细胞内基质,优先激活ERK1/2信号通路促进血管的新生[51].在细胞-细胞接触时,KLF2是Tie2诱导的促进血管静止的因素之一.Ang-1/Tie2介导的KLF2激活依赖于PI3K/Akt 途径,可反过来激活MEF2的转录活性,即KLF2通过Ang-1/Tie2间接参与血管静止与新生的调节[52].
来源骨髓的促血管生成细胞(proangiogenic cells,PACS)也称为内皮祖细胞(endothelial progenitor cells,EPCs)循环于血液中,能够促进血管新生.KLF2高表达时,在体外可使PACS 的数量增加60%,间接促进血管新生;在体内小鼠后肢缺血模型可提高老年小鼠的血管新生能力[53].
5.4 瓣膜发育
在脊椎动物中,从房室管内产生的心内膜垫形成心脏瓣膜.心内膜垫的形成过程开始于房室管的心内膜细胞,结束于上皮向间质的转化(epithelial-to-mesenchymal transformation,EMT).心内膜垫接着形成AV复合物,经过进一步转化成为功能瓣[54],表明KLF2在EMT中起重要作用的.
功能性瓣膜形成前,心房和心室之间存在顺行和逆行血流.随着成熟瓣膜的形成,顺流才成为可能.逆行流量与心动周期长度比例的变化实验即所谓的反流分数(retrograde flow fraction,RFF)表明降低RFF与斑马鱼瓣膜形成缺陷有关[55].在斑马鱼心脏房室管KLF2被逆流上调,相反,当RFF降低时KLF2下调.与在斑马鱼观察到的RFF降低出现瓣膜缺陷类似,KLF2敲除也出现瓣膜缺陷,表明KLF2及其靶基因notch1b、bmp4、edn1和nrg1在血流依赖性瓣膜形成过程中的重要作用[55].
另外,KLF2可能参与心脏瓣膜形成的证据,一种称为bungee的有缺陷心内膜垫的斑马鱼突变体,其蛋白激酶2(protein kinase 2,pkd2)失活导致HDAC5磷酸化障碍.因此,HDAC5仍处于激活状态,可结合到KLF2启动子,抑制房室区域KLF2的表达,从而降低notch1b,增加缺陷瓣膜的形成[56].
5.5 肺部发育
KLF2也被确定为正常肺发育所必需的因子.如KLF2基因敲除会导致胚胎致死, 对缺乏KLF2纯合子胚胎干细胞的嵌合体小鼠进行研究,确定其是否能够产生正常肺组织,出生后存活的小鼠,其胚胎干细胞可发育为除了肺之外的所有主要器官.相反,对出生后死亡的小鼠进行病理组织学观察,小鼠肺发育异常,表明KLF2为正常肺发育所必需的因子[57-58].
综上,KLF2不仅可维持血管稳态而且在炎性动脉粥样硬化和抗动脉粥样硬化内皮表型之间扮演了“分子开关”的角色,由于激活血管保护性程序和抑制内皮致动脉粥样硬化转录过程.KLF2可能是通过介导淋巴细胞和单核细胞/巨噬细胞生物学效应来发挥抗动脉粥样硬化作用.KLF2在胚胎和成人的生理和病理条件下的血管生成和血管新生过程中具有重要的作用,在胚胎心脏瓣膜形成中起着关键的作用.他汀类药物的多效性通过KLF2发挥作用.研究KLF2表达与血管生理机制,在心血管疾病或癌症的新疗法方面具有重要意义.
