Rosmarinic acid elicits neuroprotection in ischemic stroke via Nrf2 and heme oxygenase 1 signaling
2018-10-22HaiYingCuiXiangJianZhangYiYangCongZhangChunHuaZhuJiangYongMiaoRongChen
Hai-Ying Cui , Xiang-Jian Zhang , , Yi Yang , Cong Zhang , Chun-Hua Zhu , Jiang-Yong Miao , Rong Chen
1 Department of Neurology, Second Hospital of Hebei Medical University, Shijiazhuang, Hebei Province, China
2 Hebei Key Laboratory of Vascular Homeostasis and Hebei Collaborative Innovation Center for Cardiocerebrovascular Disease, Shijiazhuang,Hebei Province, China
Abstract
Abstract Rosmarinic acid (RA) can elicit a neuroprotective effect against ischemic stroke, but the precise molecular mechanism remains poorly understood. In this study, an experimental ischemic stroke model was established in CD-1 mice (Beijing Vital River Laboratory Animal Technology, Beijing, China) by occluding the right middle cerebral artery for 1 hour and allowing reperfusion for 24 hours. After intraperitoneally injecting model mice with 10, 20, or 40 mg/kg RA, functional neurological deficits were evaluated using modified Longa scores.Subsequently, cerebral infarct volume was measured using TTC staining and ischemic brain tissue was examined for cell apoptosis with TUNEL staining. Superoxide dismutase activity and malondialdehyde levels were measured by spectrophometry. Expression of heme oxygenase-1 (HO-1), nuclear factor erythroid 2-related factor 2 (Nrf2), Bcl-2, Bax, Akt, and phospho-Ser473 Akt proteins in ischemic brain tissue was detected by western blot, while mRNA levels of Nrf2, HO-1, Bcl-2, and Bax were analyzed using real time quantitative PCR. In addition, HO-1 enzyme activity was measured spectrophotometrically. RA (20 and 40 mg/kg) greatly improved neurological function, reduced infarct volume, decreased cell apoptosis, upregulated Bcl-2 protein and mRNA expression, downregulated Bax protein and mRNA expression, increased HO-1 and Nrf2 protein and mRNA expression, increased superoxide dismutase activity, and decreased malondialdehyde levels in ischemic brain tissue of model mice. However, intraperitoneal injection of a HO-1 inhibitor (10 mg/kg zinc protoporphyrin IX) reversed the neuroprotective effects of RA on HO-1 enzyme activity and Bcl-2 and Bax protein expression. The PI3K/Akt signaling pathway inhibitor LY294002 (10 mM) inhibited Akt phosphorylation, as well as Nrf2 and HO-1 expression. Ourfindings suggest that RA has anti-oxidative and anti-apoptotic properties that protect against ischemic stroke by a mechanism involving upregulation of Nrf2 and HO-1 expression via the PI3K/Akt signaling pathway.
Key Words: cerebral ischemia/reperfusion; rosmarinic acid; cellular apoptosis; oxidative injury; neuroprotection; Bcl-2; Bax; Nrf2; heme oxygenase 1; PI3K/Akt signal pathway; neural regeneration
Introduction
Stroke is a serious threat to human health worldwide, with ischemic stroke being the most common form. Cerebral ischemia begins with an interruption of blood flow to the brain, which is accompanied by the instant disturbance of brain tissue energy supply (Kim et al., 2017). Recombinant tissue plasminogen activator, which targets the occlusion to induce reperfusion, is currently the only drug for ischemic stroke treatment approved by the United States Food and Drug Administration (FDA) (Boese et al., 2018). However,recanalization following ischemia paradoxically exacerbates tissue damage, primarily because large amounts of reactive oxygen species (ROS) are generated upon reperfusion (Niizuma et al., 2010). ROS can attack almost all cell structures and molecules (for example, by lipid peroxidation, protein oxidation, or DNA base modification), resulting in varying degrees of functional damage to these molecules (Radak et al., 2011). Accumulating evidence has also demonstrated that generation of ROS after cerebral ischemia impairs normal mitochondrial function to result in neuronal apoptosis through activation of intrinsic apoptotic pathways (Niizuma et al., 2010). Post-ischemic oxidative damage and neuronal apoptosis play important roles in tissue loss and neurological deficits (Zhang et al., 2018). Accordingly, drugs possessing antioxidant and anti-apoptotic effects are considered beneficial for treatment of ischemic stroke.
Rosmarinic acid (α-o-caffeoyl-3,4-dihydroxyphenyl lactic acid; RA), a polyphenolic phytochemical, is commonly found in various plant families, such as Lamiaceae herbs,Boraginaceae, and the fern family Blechnaceae (Petersen and Simmonds, 2003). RA has a broad range of applications in addition to its medicinal value, and is used extensively in the food industry as aflavoring and preservative. The molecular formula of RA is C18H16O8and its structure is shown in Figure 1. This compound has many pharmacological functions including antioxidant, anti-apoptotic, antiviral, and immunomodulatory properties, yielding strong therapeutic potential for different diseases (Chu et al., 2012; Ozturk et al., 2014; Liang et al., 2016; Rahbardar et al., 2018). As shown in some preclinical studies of neurodegenerative diseases, RA has powerful neuroprotective effects in the central nervous system for Parkinson’s disease (PD), Alzheimer’s disease, and amyotrophic lateral sclerosis (Wang et al., 2012;Bigford and Del Rossi, 2014; Seo et al., 2015). Recent studies also showed that RA can protect brain tissue from ischemic damage (Luan et al., 2013; Fonteles et al., 2016; Zhang et al., 2017). However, precise roles and underlying molecular mechanisms of RA during ischemia remain unclear.
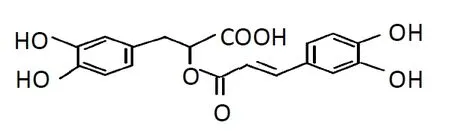
Figure 1 The chemical structure of rosmarinic acid.
Nuclear factor erythroid 2-related factor 2 (Nrf2), a member of the Cap “n” collar (CNC) family of transcription factors, is redox-sensitive and plays a very important role in cellular oxidative stress reactions. Nrf2 binds to antioxidant response elements (ARE) to upregulate expression of downstream genes encoding antioxidant proteins and phase II detoxification enzymes, such as heme oxygenase-1(HO-1), glutathione S-transferases (GSTs), and NAD(P)H quinone oxidoreductase (Jiang et al., 2017). HO-1 is the rate-limiting enzyme for heme catabolism, which results in the production of carbon monoxide (CO), iron, and biliverdin. Multiple forms of stress can activate HO-1, which is thought to afford cytoprotective effects by maintaining antioxidant/oxidant homeostasis. Indeed, in addition to the remarkable antioxidant role, induction of HO-1 also exhibited anti-apoptotic and anti-inflammatory abilities (Pae et al., 2008). Previous studies in vivo also have demonstrated that administrating some drugs which up-regulated expression of Nrf2 and HO-1 could reduce oxidative damage and protect brain against ischemic injury (Yang et al., 2009; Li et al., 2011; Chen et al., 2012).
This study investigated the neuroprotective effects of RA in a mouse model of ischemic stroke [achieved by 1-hour occlusion of the middle cerebral artery (MCA)], and examined whether such effects were related to attenuation of apoptotic cell death and elevation of Nrf2/HO-1 expression.In addition, this study investigated the role of RA-induced HO-1 in reducing apoptosis following ischemic stroke, as well as the involvement of PI3K/Akt signaling in regulation of Nrf2 and HO-1 expression after RA administration.
Materials and Methods
Experimental ischemic stroke model establishment
Experimental procedures were in line with the National Institutes of Health Guide for Care and Use of Laboratory Animals, and approved by the Committee of Experimental Animal Administration of Hebei Medical University (approval No. 2015216). One-hundred forty-one male specific pathogen free CD-1 mice [Vital River Company, Beijing, China,License No. SCXK (Jing) 2012-0001], weighing 25–30 g and aged 4 weeks, underwent focal transient cerebral ischemia as previously described (Yang et al., 2009). The right MCA was occluded for 1 hour and then re-perfused for 24 hours. Sham-operated mice underwent the same surgical procedure withoutfilament insertion. All mice were provided with free access to food and water, and were allowed at least 2 days to adapt to new environments without surgery.
Groups and drug administration
Rosmarinic acid (RA; ZeLang Biotechnology, Nanjing,China) with a purity of 98% was dissolved in saline. Upon reperfusion, RA was immediately injected intraperitoneally at dosages of 10, 20, or 40 mg/kg according to a previously reported study of RA in humans (Noguchi-Shinohara et al.,2015). Zinc protoporphyrin IX (ZnPPIX, a specific HO-1 inhibitor, 10 mg/kg; Sigma-Aldrich, St. Louis, MO, USA) was administered intraperitoneally 24 hours before ischemia.ZnPPIX was prepared by dissolving in 2 mL of 0.2 M NaOH,then adjusting the pH value to 7.4 using 1 M HCl and diluting to afinal volume with 0.9% NaCl. LY294002 (PI3K/Akt pathway inhibitor, 10 mM; Sigma-Aldrich) was dissolved in 2% dimethyl sulfoxide (DMSO) for administration (i.c.v.,5 μL; bregma: 0.9-mm lateral, 1.5-mm posterior, 3.5-mm deep) 15 minutes before ischemia.
Experiment 1: All mice were randomly divided into five groups (n = 18 per group): sham, transient middle cerebral artery occlusion (tMCAO), RA-L (10 mg/kg RA + tMCAO),RA-M (20 mg/kg RA + tMCAO), and RA-H (40 mg/kg RA+ tMCAO). Mice in the tMCAO group received an equal volume of 0.9% saline intraperitoneally.
Experiment 2: All mice were randomly divided into four groups (n = 9 per group): sham, tMCAO, RA (40 mg/kg RA+ tMCAO), and RA + ZnPPIX (40 mg/kg RA + ZnPPIX +tMCAO). Mice in the tMCAO group received an equal volume of 0.9% saline.
Experiment 3: All mice were randomly divided into five groups (n = 3 per group): sham, tMCAO, RA (40 mg/kg +tMCAO), RA + LY (40 mg/kg RA + LY294002 + tMCAO),and DMSO (DMSO + tMCAO). Mice in the DMSO group received tMCAO and an equal volume of 2% DMSO.
Neurological function assessment
An investigator blinded to the experimental design carried out neurological testing 24 hours after reperfusion (n = 10 per group). Deficits were scored according to a modified Longa scoring system as follows: 0, no deficits; 1, difficulty in fully extending the contralateral forelimb; 2, unable to extend the contralateral forelimb; 3, mild circling to the contralateral side; 4, severe circling; and 5, falling to the contralateral side (Longa et al., 1989).
2,3,5-Triphenyltetrazolium chloride (TTC) staining
Twenty-four hours after reperfusion, brains from mice (n =6 per group) were cut into 2-mm thick slices and incubated in a 2% TTC solution at 37°C for 15 minutes (Bederson et al., 1986). Stained sections were photographed and digital images were analyzed using an Image Pro-Plus5.1 analysis system (Media Cybernetics, Rockville, MD, USA). Lesion volumes were calculated by the following equation (Tatlisumak et al., 1998): percent hemispheric lesion volume(%HLV) = {[total infarct volume − (right hemisphere volume – left hemisphere volume)]/left hemisphere volume}× 100%. An additional examiner blinded to experimental groupings measured the infarct volume.
TUNEL staining
Twenty-four hours after reperfusion, mice (n = 3 per group)were anesthetized and perfused through the heart with cold saline followed by 4% paraformaldehyde in phosphate buffer. Removed brains were fixed in 4% paraformaldehyde overnight and then embedded in paraffin. Brain tissues were cut into 5-μm thick coronal sections, deparaffinized, and rehydrated. For TUNEL staining, sections were processed with an In Situ Cell Death Detection Kit, AP (Roche, Diagnostics GmbH, Mannheim, Germany). TUNEL-positive cells were visualized by light microscopy and quantified infive non-overlapping visualfields for each slice.
Determination of superoxide dismutase (SOD) and malondialdehyde (MDA) levels
Ischemic brain tissues were collected 24 hours after reperfusion (n = 3 per group). Samples were rinsed, weighed,and homogenized in 9 volumes of 9 g/L ice-cold saline for 10 minutes using a Dounce Tissue Grinder (Kimble and Kontes,Vineland, NJ, USA). After centrifugation at 1430 × g for 10 minutes at 4°C, the homogenate supernatant was obtained.SOD activity was detected using a Total SOD (T-SOD)Assay kit (hydroxylamine method) (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) based on the production of superoxide ions by a xanthine-xanthine oxidase system, which can be measured at 550 nm. One unit (U) was defined as the protein amount that inhibited the system by 50%. MDA was examined based on the absorbance at 532 nm of thiobarbituric acid-reactive substances using an MDA assay kit (Nanjing Jiancheng Bioengineering Institute).
Western blot assay
Ischemic brain tissues were collected 24 hours after reperfusion. Nuclear Nrf2 protein and total protein levels of HO-1, Bcl-2, and Bax were measured using a Nuclear-Cytosol-Mem Extraction kit (Applygen Technologies, Beijing,China). Total protein concentrations were measured with a BCA Protein Assay kit (Novagen, Madison, WI, USA). Equal amounts of protein samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. Membranes loaded with various proteins were incubated in blocking buffer (5% non-fat dried milk in phosphate-buffered saline) at room temperature for 2 hours and then incubated overnight at 4°C with the corresponding primary antibodies: mouse monoclonal anti-HO-1 (1:500; Enzo Biochem,Farmingdale, NY, USA), rabbit polyclonal anti-Nrf2 (1:200;Santa Cruz Biotechnology, Inc, Dallas, TX, USA), rabbit polyclonal anti-Bcl-2 (1:500; Bioworld Technology, St. Louis Park, MN, USA), rabbit polyclonal anti-Bax (1:500; Bioworld Technology), rabbit polyclonal anti-phospho-Ser473 Akt (1:500; Cell Signaling Technology, Danvers, MA, USA),and rabbit polyclonal anti-Akt (1:500; Cell Signaling Technology). The following day, membranes loaded with primary antibodies were washed with 0.1% Tween-20 Tris-buff-ered saline and then incubated with secondary antibodies(goat anti-rabbit/mouse, 1:5000; Rockland, Gilbertsville,PA, USA) for 1 hour at room temperature. Anti-mouse β-actin (1:500; Zhongshan Biotechnology, Beijing, China)was used as an internal control (n = 3 per group). Antibody-antigen complexes were detected using an enhanced chemiluminescence western blotting detection kit (Thermo Fisher Scientific, Waltham, MA, USA) and Light-Capture system (Bio-Rad ChemiDoc XRS+, Hercules, CA, USA).Optical density values of protein bands were determined by ImageJ (National Institutes of Health, Bethesda, MD, USA)and normalized to β-actin.
Quantitative real-time polymerase chain reaction(qRT-PCR)
Ischemic brain tissues were collected 24 hours after reperfusion. mRNA detection of Nrf2, HO-1, Bcl-2, and Bax was carried out by qRT-PCR (n = 3 per group). Total RNA was extracted from frozen ischemic or sham-operated hemispheres using Trizol reagent. Reverse transcription was carried out using afirst-strand cDNA synthesis kit (Fermentas International, Burlington, ON, Canada). Obtained cDNA was amplified by a real-time PCR system (Agilent, Santa Clara, CA, USA) in the presence of afluorescent dye (SYBR Green I; CWBIO, Beijing, China). Relative abundance of mRNA was calculated after normalization to β-actin mRNA.Forward and reverse primers are listed in Table 1.

Table 1 Summary of the quantitative real-time polymerase chain reaction primer sequences
HO-1 enzymatic activity assay
HO-1 enzymatic activity was measured by spectrophotometric determination of bilirubin formation (n = 6 per group)as previously described (Shih et al., 2010). Briefly, brain tissues were homogenized and centrifuged at 15,000 × g for 40 minutes at 4°C. Supernatants were acquired and mixed with buffer (1 mM glucose 6-phosphate, 1 U/mL glucose-6-phosphate dehydrogenase, 0.5 mM NADPH, 10 μM hemin, 200 μg biliverdin reductase, 1 mM potassium phosphate buffer)for 1 hour at 37°C. Amounts of bilirubin were calculated by the difference in absorbance between 464 nm and 530 nm using a spectrophotometer (Evolution 260, Thermo Fisher).
Statistical analysis
Quantitative data are expressed as the mean ± SD. Statistical analysis was performed with SPSS 13.0 software (SPSS, Chicago,IL, USA). Statistical comparisons were conducted using oneway analysis of variance followed by a least significant difference test for multiple comparisons. The Mann-Whitney U test was used for comparison of neurological deficit scores between two groups. Differences were considered significant if P < 0.05.
Results
Experiment 1
RA attenuates ischemic brain injury after tMCAO
Neurological deficit scores and infarct volumes were examined to detect whether RA decreased brain damage after tMCAO. With the exception of mice in the sham group, all animals presented significant functional neurological deficits 24 hours after reperfusion. Neurological deficit scores were significantly decreased in RA-M and RA-H groups compared with the tMCAO group (P < 0.05; Figure 2A),however there was no significant difference in neurological deficit scores between RA-L and tMCAO groups (P > 0.05;Figure 2A).
TTC staining can distinguish infarcted from normal brain tissue. The sham group showed no infarction. However,the tMCAO group had a large infarct in the ischemic hemisphere. Infarct volume was obviously reduced in RA-M and RA-H groups compared with the tMCAO group (P < 0.01;Figure 2B and C).
According to the observations described above, RA at middle and high dosages (20 and 40 mg/kg, respectively)provided better protection after tMCAO. Therefore, we focused on these two dosages in our subsequent study of the molecular mechanism by which RA protects the brain against ischemic injury.
RA alleviates apoptosis after tMCAO
The results of TUNEL staining to detect apoptotic cells are shown in Figure 3A. A few cells were detected in the sham group, but there were large numbers of TUNEL-positive apoptotic cells in ischemic cerebral hemispheres of the tMCAO group. RA-M and RA-H significantly reduced the numbers of TUNEL-positive cells (P < 0.01, vs. tMCAO group), although this effect was more pronounced in the RA-H group (P < 0.05, vs. RA-M group; Figure 3B).
To further investigate potential neuroprotective mechanisms of RA for alleviating apoptosis after tMCAO, expression of two important apoptosis-related proteins, Bcl-2 and Bax, was analyzed. Compared with the sham group, western blot results showed a significant decrease in Bcl-2 and obvious increase in Bax in the tMCAO group (P < 0.01; Figure 3C and D). However, RA-H and RA-M significantly increased Bcl-2 protein expression and decreased Bax protein expression (P < 0.05 or P < 0.01; Figure 3C and D). Changes in Bcl-2 and Bax protein expression were more pronounced in the RA-H group than the RA-M group (P < 0.05 or P <0.01; Figure 3C and D). qRT-PCR results indicated that changes in mRNA expression in different groups were similar to those observed by western blot assay (Figure 3E and F).
RA attenuates oxidative stress after tMCAO
Cortical SOD activity and MDA levels in different groups were evaluated 24 hours after tMCAO. As shown in Table 2, SOD activity was significantly increased and MDA levels were significantly decreased in the RA-M and RA-H groups compared with the tMCAO group (P < 0.05). This inhibitory effect was stronger in the RA-H group than the RA-M group(P < 0.05).
RA increases nuclear Nrf2 and HO-1 expression
Protein and gene levels of Nrf2 and HO-1 were examined by western blot assay and qRT-PCR (Figure 4). Small amounts ofnuclear Nrf2 and HO-1 protein were expressed in sham group mice, while levels were increased 24 hours after reperfusion in the tMCAO group (P < 0.01; Figure 4A and B). As expected,Nrf2 and HO-1 protein expression in RA-M and RA-H groups was significantly upregulated (P < 0.01; Figure 4A and B).Moreover, Nrf2 and HO-1 protein levels were significantly upregulated in the RA-H group compared with the RA-M group(P < 0.01; Figure 4A and B). qRT-PCR results also demonstrated that RA-M and RA-H increased gene expression of nuclear Nrf2 and HO-1 (P < 0.05 or P < 0.01; Figure 4C and D).

Table 2 Effect of RA on MDA (nmol/mg) and SOD (U/mg) levels in ischemic brain tissues of tMCAO mice 24 hours after reperfusion
Experiment 2
Anti-apoptotic ability of RA is inhibited by ZnppIX
To further investigate the anti-apoptotic role of HO-1 induced by RA treatment, the HO-1 activity inhibitor ZnPPIX was used in combination with high dose RA to identify underlying mechanisms. Changes in HO-1 enzymatic activity was detected in different groups. As shown in Figure 5A,HO-1 activity increased after ischemia, and was elevated in RA-treated mice. HO-1 activity was significantly reduced in the RA + ZnPPIX group compared with the RA group (P <0.01; Figure 5A). Western blot results showed decreased Bax and increased Bcl-2 in the RA group, which were reversed by ZnppIX. Moreover, there were significant differences in Bax and Bcl-2 expression between RA and RA + ZnppIX groups (P < 0.05 or P < 0.01; Figure 5C and D).
Experiment 3
LY294002 inhibits p-Akt and suppresses RA-induced Nrf2 and HO-1 expression after tMCAO
To examine the role of PI3K/Akt signaling in regulation of RA-induced Nrf2 and HO-1 after tMCAO, the compound LY294002 was used to inhibit the PI3K/Akt signaling pathway. Western blot analysis showed increased HO-1 and Nrf2 (Figure 6B and D) and decreased p-Akt (Figure 6C) in ischemic hemispheres after 24 hours of reperfusion in tMCAO and DMSO groups. p-Akt expression was significantly upregulated by RA treatment and this effect was reduced by LY294002 administration 15 minutes prior to ischemia (P< 0.01 or P < 0.05; Figure 6A and C). Protein levels of Akt were unaffected. Co-administration of LY294002 inhibited RA-induced increases of Nrf2 and HO-1, as indicated by statistically significant differences between RA and RA+LY groups (P < 0.01; Figure 6B and D).
Discussion
tMCAO is a classic model of cerebral ischemia that is commonly used to investigate mechanisms of ischemic stroke and evaluate neuroprotective drugs (Yang et al., 2009; Liu et al., 2014). In our study, we investigated the therapeutic role of RA in ischemia/reperfusion brain injury and potential underlying mechanisms. Our results showed that RA improved neurological dysfunction; decreased infarct volume; reduced TUNEL-positive cells; upregulated Bcl-2, HO-1, Nrf2, and SOD expression; and significantly downregulated Bax and MDA expression. Moreover, the PI3K/Akt pathway was involved in the upregulation of Nrf2 and HO-1 induced by RA.
The occurrence of ischemic stroke begins with a decrease in blood supply that is accompanied by multiple detrimental processes including ROS overproduction, consumption of antioxidants, and inactivation of detoxification systems.These changes disrupt the normal anti-oxidative defense capacity of brain tissue (Chan, 2001). MDA, a toxicfinal product of lipid peroxidation, promotes cross-linking of nucleic acids, proteins, and phospholipids, which results in dysfunction of biological macromolecules (Yuan et al., 2017).MDA levels can reflect the degree of lipid peroxidation and act as a key marker of oxidative stress (Schettler et al., 2009).SOD, an endogenous antioxidant enzyme, converts harmful superoxide radicals into hydrogen peroxide, thereby serving as a superoxide radical scavenger to provide cytoprotection against damage induced by toxic oxygen free radicals(Ikonomidou and Kaindl, 2011; Chen et al., 2017). We measured MDA levels and SOD activity in mice with ischemic stroke after RA treatment. RA increased SOD activity and decreased MDA levels in ischemic brain tissue 24 hours after tMCAO, suggesting RA can scavenge ROS instantly by increasing endogenous antioxidant enzyme activity and reducing lipid peroxidation.
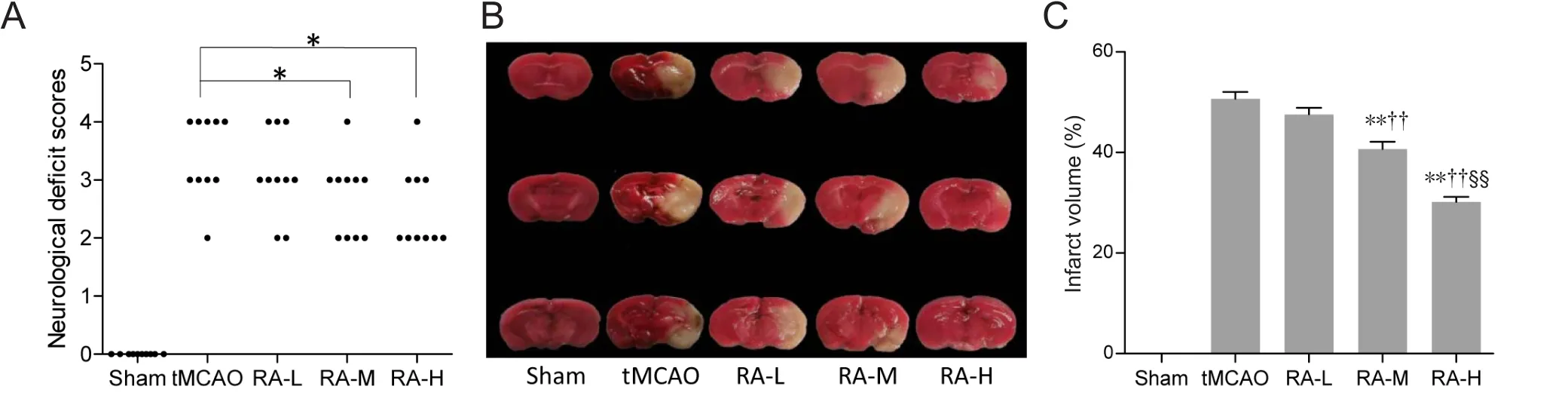
Figure 2 Rosmarinic acid (RA) improves neurological outcomes 24 hours after reperfusion.
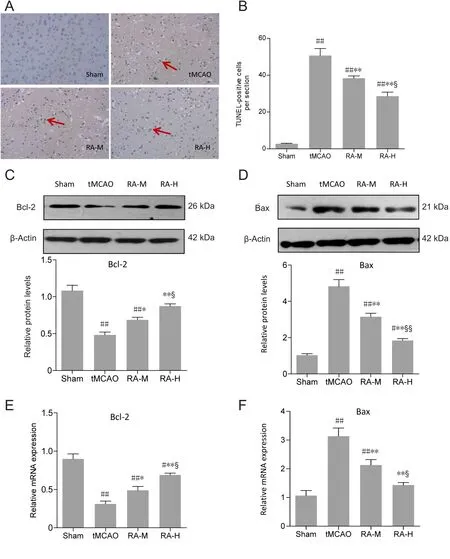
Figure 3 Rosmarinic acid (RA)alleviates apoptosis after tMCAO after 24 hours of reperfusion.
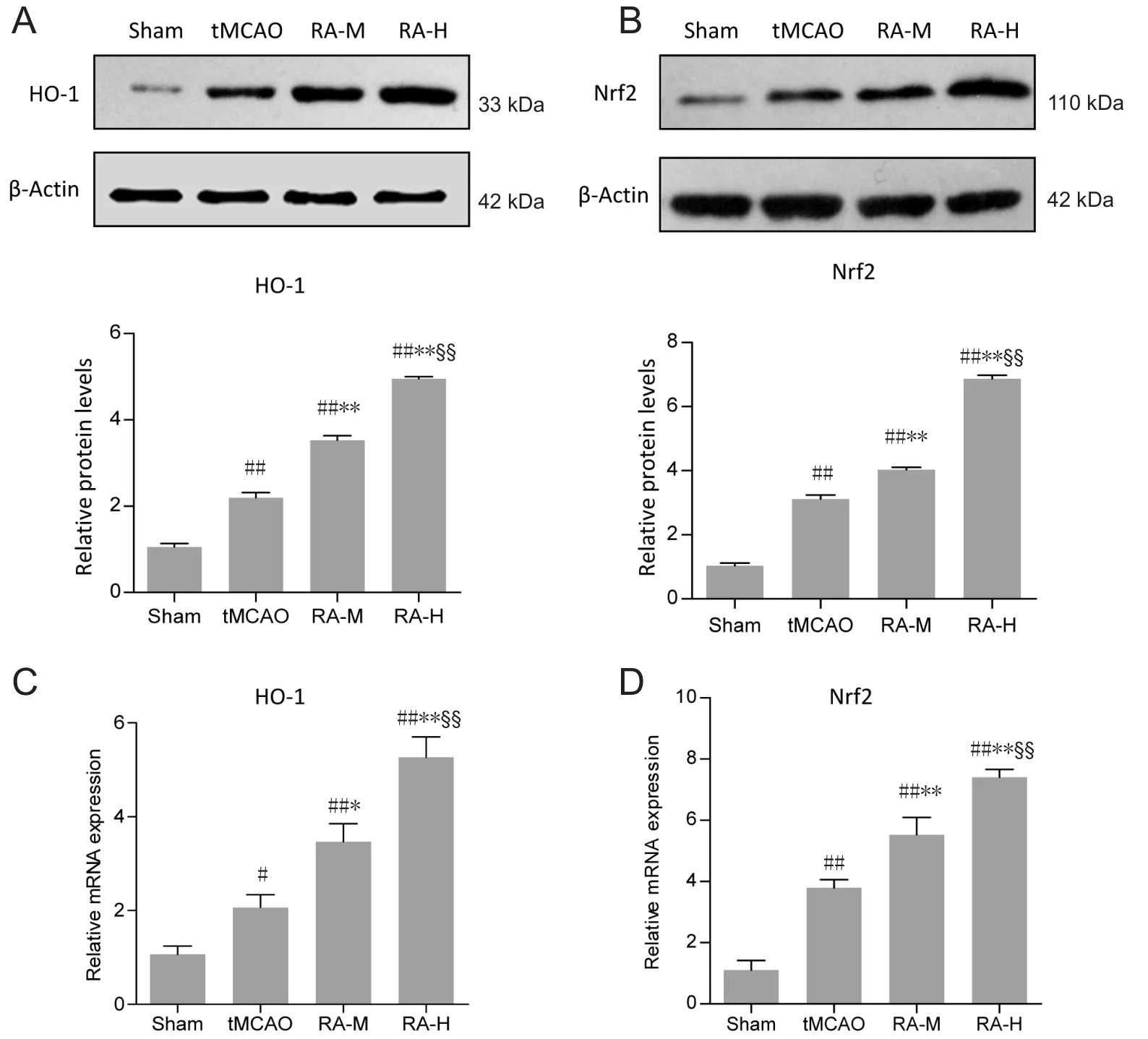
Figure 4 RA increases protein and mRNA expression of Nrf2 and HO-1 in ischemic brain tissues 24 hours after reperfusion.
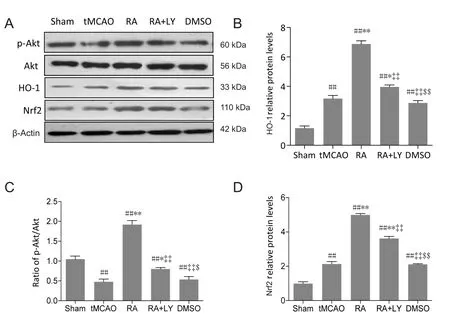
Figure 6 LY294002 (LY), a PI3K/Akt pathway inhibitor, inhibites p-Akt and attenuates RA-induced upregulation of Nrf2 and HO-1 in ischemic brain tissues 24 hours after reperfusion.
Nrf2 is a key endogenous regulator for defending against oxidative stress. Under normal physiological conditions,Nrf2 binds to kelch-like ECH-related protein 1 (Keap1)in the cytoplasm and is continuously ubiquitinated and degraded, thus rendering Nrf2 unable to enter the nucleus and exert transcriptional activity. Upon stimulation, Nrf2 is activated by dissociation from Keap1, thus permitting Nrf2 to enter the nucleus and trigger expression of a battery of genes encoding numerous protective phase II defense enzymes to restore cellular redox homeostasis (Jiang et al.,2017). Several preclinical studies have revealed that activating the Nrf2/ARE pathway affords cytoprotection against oxidative stress and metabolic disorders both in vitro and in vivo (Satoh et al., 2008; Yang et al., 2016b). This feature is particularly prominent in many central nervous system disease models, such as cerebral ischemia and PD. Moreover, primary cultured neurons from Nrf2-deficient mice were shown to be more sensitive to oxidative injury, calcium influx, and mitochondrial toxicity than cells derived from wild-type mice (Lee et al., 2003). A recent study using obese Nrf2+/+and Nrf2–/–mice reported that progressive Nrf2 dysfunction played a role in the aging process, and age-related Nrf2 dysfunction participated in pathological mechanisms of vascular cognitive impairment (Tarantini et al., 2018).Our previous findings also suggested that Nrf2-knockout mice were more vulnerable to oxidative stress (Li et al.,2013). As an important downstream enzyme of the Nrf2/ARE signaling pathway, HO-1 has been highlighted for its effective neuroprotection against ischemic injury and neurodegenerative diseases (Yang et al., 2009; Li et al., 2015).Indeed, HO-knockout mice exhibit exacerbated cerebral infarcts (Saleem et al., 2008), whereas HO-1 overexpression reduced brain injury (Yang et al., 2016c). Notably, although HO-1 is redox-sensitive, it can be induced by various forms of stimuli. HO-1 exerts an indirect antioxidant effect primarily by degrading heme to CO and biliverdin/bilirubin,which possess powerful antioxidant functions (Pae et al.,2008). Consequently, the induction of HO-1 through Nrf2 activation represents a potential therapeutic approach for ischemic stroke. Our present study demonstrated significantly upregulated expression of nuclear Nrf2 and HO-1 in ischemic cortex after RA administration at 24 hours after tMCAO, indicating a stronger activation of cell stress responses against ischemia/reperfusion brain injury in mice.Moreover, these results indicated that activation of the Nrf2/ARE/HO-1 pathway during the early stage may be one potential mechanism by which RA is neuroprotective against ischemic injury. A recent study showed that RA activated Nrf2/HO-1 and ameliorated acute liver damage induced by CCl4 (Domitrović et al., 2013), further supporting our results. Furthermore, the noise-activated Nrf2/HO-1 pathway was enhanced by RA, thus protecting cochlea against noise-induced injury (Fetoni et al., 2015).
Neuronal apoptosis is recognized as a prominent feature in pathological processes; indeed, inhibition of apoptosis and related cell death pathways can reduce ischemia/reperfusion brain injury (Niizuma et al., 2010). In our study,treatment with RA enhanced resistance to cell apoptosis during ischemic stroke, as demonstrated by reduced numbers of TUNEL-positive cells, decreased Bax, and markedly increased Bcl-2. Bax and Bcl-2, which belong to a large family of Bcl-2-like proteins, play significant roles in regulating apoptosis after ischemic stroke. Bcl-2, an anti-apoptotic molecule, is localized to the mitochondrial membrane, whereby it acts as a gatekeeper to maintain membrane integrity (Roy et al., 2014). In contrast, Bax, a pro-apoptotic molecule, leads to permeabilization of the outer mitochondrial membrane and acceleration of cytochrome c release. The formation of Bcl-2/Bax heterodimers can weaken the apoptosis-promoting effect of Bax and affect intracellular apoptosis signal transduction. In a model of focal ischemic injury, the infarct volume of transgenic mice overexpressing Bcl-2 in neurons was half that of wild-type mice (Martinou et al., 1994). As reported previously, RA can inhibit apoptosis of cardiac muscle cells (Kim et al., 2005; Iuvone et al., 2006) and rescued neuronal activity in a rat model of PD by changing the Bcl-2/Bax ratio (Wang et al., 2012). Consistent with these reports,our present observations demonstrated that RA protected neurons against apoptosis induced by ischemic insult.
Promoting HO-1 expression with various pharmacological agents has been shown to prevent apoptosis in several preclinical models of tissue injury (Vulapalli et al., 2002;He et al., 2014). Vulapalli et al. (2002) reported that cardioselective overexpression of HO-1 attenuated apoptosis in mouse heart after myocardial ischemia/reperfusion. Recently, He et al. (2014) showed that pharmaceutical induction of HO-1 reduced retinal cell apoptosis and provided protection against retinal ischemia injury. Elevated HO-1 and its enzymatic byproducts impart an anti-apoptotic effect by regulating apoptotic pathway proteins. In addition, HO-1 overexpression can reportedly influence mitochondrial transport carriers and functions by activating anti-apoptotic molecules such as BcL-XL and inhibiting cytochrome c release(Di Noia et al., 2006). Biliverdin therapy has been shown to downregulate pro-apoptotic molecules (cytochrome c and caspase-3) and reduce reperfusion damage in rat liver grafts(Tang et al., 2007). CO inhibits the release of caspases (-3,-8, -9) and mitochondrial cytochrome c (Zhang et al., 2003)to exert anti-apoptotic effects. These studies aroused our interest in detecting the role of RA-mediated HO-1 elevation in alleviating apoptosis after tMCAO. Importantly, the protective effect of RA on regulating Bax and Bcl-2 expression was reversed by the HO-1 activity inhibitor ZnPPIX. Therefore, we concluded that RA conferred cytoprotection against apoptosis in ischemic stroke by increasing HO-1 expression and enhancing HO-1 activity.
As previously reported, the PI3K/Akt pathway (which is involved in cell growth, proliferation, and differentiation across a broad variety of cells) can be activated under various physiological and stress stimuli such as growth factors,hypoxia, and oxidative stress in various tissues, and is a key component of protective mechanisms against ischemic stroke(Chien et al., 2016; Yang et al., 2016a; Lee et al., 2017). Once Akt is activated, it can phosphorylate many targets to affect multiple cellular processes relating to cell growth, proliferation, or apoptosis. Activated Akt can also promote Nrf2 nuclear translocation (Huang et al., 2002; Franke et al., 2003). In our study, RA significantly increased p-Akt, Nrf2, and HO-1, whereas co-administration with LY294002 (a PI3K/Akt pathway inhibitor) hindered upregulation of Nrf2 and HO-1.These results demonstrated a positive correlation between the PI3K/Akt pathway and RA-induced upregulation of Nrf2 and HO-1. Therefore, we concluded that activating the Akt/Nrf2/HO-1 pathway provides a valuable therapeutic target for RA to protect the brain against acute ischemic damage.
In summary, systemic administration of RA exerted protective effects against ischemia/reperfusion injury in brain tissue via anti-oxidative and anti-apoptotic properties,which could be attributed, at least in part, to elevation of Nrf2 and HO-1 via the PI3K/Akt pathway. However, our preclinical study results and experimental design have some limitations. Although tMCAO is a classic model of cerebral ischemia, it cannot represent all clinical ischemic stroke types. Moreover, the mice used in our study were healthy male mice that did not suffer from atherosclerosis prior to modeling, which is not completely consistent with common stroke patients. Regardless, RA is a promising neuroprotective drug and more research is needed to establish its potential for clinical application.
Acknowledgments:We thank Rui-Chun Liu (Second Hospital of Hebei Medical University, China) and Hong-Ran Wu (Second Hospital of Hebei Medical University) for their technical assistance and Prof. Yan-Su Guo, MD, PhD (Second Hospital of Hebei Medical University) and Wei-Song Duan (MD, PhD, Second Hospital of Hebei Medical University) for providing valuable suggestions.
Author contributions:Study design: HYC and XJZ; animal model establishment and TUNEL staining conduction: HYC and YY; PCR implement and western blot analysis: CZ, CHZ, JYM, and RC; data analysis andfigure preparation: HYC and YY; paper writing: HYC, XJZ, YY, CZ,CHZ, JYM and RC. All authors approved thefinal version of this paper for publication.
Conflicts of interest:No conflicts of interest exit in the submission and publication of this manuscript.
Financial support:This work was generously supported by the National Natural Science Foundation of China, No. 81571292 (to XJZ), and No.81601152 (to YY); the Natural Science Foundation of Hebei Province of China, No. H2017206338 (to RC). The funding bodies played no role in thestudy design, in the collection, analysis and interpretation of data, in the writing of the paper, or in the decision to submit the paper for publication
Institutional review board statement:The study was approved by the Committee of Experimental Animal Administration of Hebei Medical University (No. 2015216).
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix,tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers:Han-A Park, University of Alabama, USA.
Additionalfile:Open peer review report 1.
杂志排行
中国神经再生研究(英文版)的其它文章
- Huangqinflavonoid extraction for spinal cord injury in a rat model
- Lithium promotes recovery of neurological function after spinal cord injury by inducing autophagy
- Analysis of transcriptome sequencing of sciatic nerves in Sprague-Dawley rats of different ages
- Exogenous brain-derived neurotrophic factor attenuates cognitive impairment induced by okadaic acid in a rat model of Alzheimer’s disease
- Partial improvement in performance of patients with severe Alzheimer’s disease at an early stage of fornix deep brain stimulation
- Epigenetic marks are modulated by gender and time of the day in the hippocampi of adolescent rats:a preliminary study