Exogenous brain-derived neurotrophic factor attenuates cognitive impairment induced by okadaic acid in a rat model of Alzheimer’s disease
2018-10-22AiHuaXuYangYangYongXinSunChaoDongZhang
Ai-Hua Xu, Yang Yang, Yong-Xin Sun, Chao-Dong Zhang
1 Department of Rehabilitation Medicine, the First Affiliated Hospital of China Medical University, Shenyang, Liaoning Province, China
2 Department of Neurology, the First Affiliated Hospital of China Medical University, Shenyang, Liaoning Province, China
Abstract Decreased expression of brain-derived neurotrophic factor (BDNF) plays an important role in the pathogenesis of Alzheimer’s disease,and a typical pathological change in Alzheimer’s disease is neurofibrillary tangles caused by hyperphosphorylation of tau. An in vivo model of Alzheimer’s disease was developed by injecting okadaic acid (2 μL) and exogenous BDNF (2 μL) into the hippocampi of adult male Wister rats. Spatial learning and memory abilities were assessed using the Morris water maze. The expression levels of protein phosphatase 2A (PP2A), PP2Ac-Yp307, p-tau (Thr231), and p-tau (Ser396/404) were detected by western blot assay. The expression levels of BDNF, TrkB, and synaptophysin mRNA were measured by quantitative real-time polymerase chain reaction. Our results indicated that BDNF expression was suppressed in the hippocampus of OA-treated rats, which resulted in learning and memory deficits. Intra-hippocampal injection of BDNF attenuated this OA-induced cognitive impairment. Finally, ourfindings indicated an involvement of the PI3K/GSK-3β/AKT pathway in the mechanism of BDNF in regulating cognitive function. These results indicate that BDNF has beneficial effect on Alzheimer’s disease, and highlight the potential of BDNF as a drug target for treatment of Alzheimer’s disease.
Key Words: nerve regeneration; Alzheimer’s disease; exogenous brain-derived neurotrophic factor; Tau protein; okadaic acid; phosphorylation;PP2A-Y307; glycogen synthase kinase-3β; TrkB; cognitive function; brain protection; neural regeneration
Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disease, accounting for 50–75% of all dementias;about 131 million people worldwide have dementia, and AD is expected to become 3-fold more prevalent by 2050(Wirz et al., 2014; Alzheimer’s Association, 2015; Purnell et al., 2015). Neurofibrillary tangles are typically seen in patients with AD and are strongly associated with dementia severity in these patients (Whittington et al., 2013). The main component of these neurofibrillary tangles is hyperphosphorylated tau protein (Liu et al., 2008; Ye et al., 2017).Tau protein normally exists in the axon and cytoplasm of neurons. The microtubule-associated protein tau stabilizes microtubules, which supports the axonal transport of proteins, vesicles, and organelles (Morris et al., 2011). The biological activity of Tau protein is regulated by phosphorylation and dephosphorylation. Protein phosphatase 2A(PP2A) is the major intracerebral phosphoesterase, which dephosphorylates the tau protein. A previous study showed that the activity and expression of PP2A were reduced by 20% in a mouse model of AD (Zhao et al., 2013). Inhibition of PP2A activity by decreasing PP2A methylation of the catalytic subunit of L309 or increasing Y307 phosphorylation may result in the aggregation and hyperphosphorylation of tau protein (Zhou et al., 2008; Xiong et al., 2013). At present,it is believed that AD and other neurodegenerative diseases are associated with impaired axonal transport, an imbalance of neurotrophic factors (Schindowski et al., 2008; Gan et al.,2015). The over-phosphorylation of Tau protein can lead to abnormal microtubule function, cytoskeleton instability,and axonal transport dysfunction (Lu et al., 2013; Metaxas and Kempf, 2016). Furthermore, it has been reported that abnormal hyperphosphorylation of tau protein is an early manifestation of AD (Caraci et al., 2013; Zhang et al., 2016);thus, inhibition of tau protein phosphorylation is thought to be the key to AD therapy (Iqbal et al., 2014).
Brain-derived neurotrophic factor (BDNF) is a neurotrophic factor that can promote the survival and growth of many kinds of neurons, increase the activity of antioxidant enzymes in cells andfight against free radicals, and inhibit the excitatory amino acid cytotoxicity and apoptosis (Huang and Reichardt, 2001; Liu et al.,2016; Sampaio et al., 2017).BDNF also participates in cognition, learning, and memory formation (Yamada and Nabeshima, 2003). The levels of BDNF and its receptor have been found to be markedly decreased in the hippocampus of patients with AD, which may cause the cognitive impairment (Danzer et al., 2004).Conversely, enhanced levels of BDNF may delay age-related cognitive decline, including AD neuropathology, which suggests that BDNF may be a biomarker for the diagnosis and treatment of AD (Beeri and Sonnen, 2016; Buchman et al., 2016). However, the underlying mechanisms by which BDNF affects AD are still not clear.
We used okadaic acid (OA) to establish an animal and cell model of AD, evaluated the beneficial role of exogenous BDNF in OA-induced hyperphosphorylation of tau and the resulting cognitive dysfunction in vivo and in vitro, and also elucidated the possible mechanisms of this effect.
Materials and Methods
Animals
A total of 36 healthy adult male Wister rats aged 7–8 weeks and weighing 200 ± 18 g were provided by the Experimental Animal Center of China Medical University, China (license number: SCXK (Liao) 2003-0001). These rats were housed at 20 ± 2°C with natural ventilation and 12-hour light-dark cycle. Animals had access to standard rat chow and water ad libitum. The present study was approved by the Animal Care and Use Committee of China Medical University (ethical approval number: 1003M) and performed in accordance with the National Institutes of Health Guidelines of the Care and Use of Laboratory Animals.
Induction of AD models in vivo and drug intervention
The animal model of AD was prepared by stereotaxic injection technique with OA (Broetto et al., 2016). Rats were anesthetized with intraperitoneal injection of 10% chloral hydrate(300 mg/kg) and mounted on a stereotaxic apparatus (RWD Life Science Co., Ltd., Shenzhen, China). The skulls of the rats were opened and drilled with burr holes on both sides of hippocampal CA3 area according to a rat brain stereotaxic atlas (anteroposterior: −3.8 mm, mediolateral: ±3.8 mm, dorsoventral: 4.0 mm) (Shirazi-Southall et al., 2002).
The 36 rats were randomly and equally divided into six groups (n = 6 per group) as follows: (1) Sham group: Both hippocampi were injected with 2 μL of artificial cerebrospinalfluid; (2) OA group: Both sides of the hippocampus were injected with 2 μL of OA (0.2 μM; Upstate Biotechnology,Inc., Lake Placid, NY, USA) dissolved in dimethyl sulfoxide at 1 μM and diluted to 0.2 μM in artificial cerebrospinalfluid; (3) OA + BDNF group: Both sides of the hippocampus were injected with 2 μL of OA (0.2 μM) and 2 μL of BDNF(50 ng/mL human full-length BDNF protein; Millipore Corp.,Billerica, MA, USA) (Yuan et al., 2017); (4) OA + BDNF +K252a group: Both sides of the hippocampus were injected with 2 μL of OA (0.2 μM), 2 μL of BDNF (50 ng/mL), and 2 μL of K252a (0.2 μM, the inhibitor of the BDNF-specific receptor, TrkB; Santa Cruz Biotechnology, Santa Cruz, CA,USA); (5) OA + BDNF + LY294002 group: Both sides of the hippocampus were injected with 2 μL of OA (0.2 μM), 2 μL of BDNF (50 ng/mL), and 2 μL of LY294002 (0.2 μM, the inhibitor of PI3K, Santa Cruz Biotechnology); (6) rats in the normal control group received no treatment. The injection lasted for 5 minutes, and the needle with the syringe was left in place for 2 minutes after the injection to ensure complete infusion of the drug. After surgery, the rats were housed individually and had free access to food and water. Penicillin was applied daily,and the rats were allowed 7 days to recover from surgery. No unintended deaths of animals occurred during the surgery.The general condition of the animals, including body weight,food and water intake, was monitored daily after surgery.
Cell culture, induction of AD model in vitro, and drug intervention
The SH-SY5Y cells (human neuroblastoma cells, derived from human neuroblastoma cell lines SK2N and 2SH) were obtained from Shanghai Institute of Cell Biology (Shanghai,China) and maintained in Dulbecco’s modified Eagle’s medium (DMEM)-F12 (Gibco, Grand Island, NY, USA) supplemented with 10% (v/v) fetal calf serum (Hyclone, Logan, UT,USA), 2 mM L glutamine (Sigma, Santa Clara, CA, USA), and 1% non essential amino acids (100× stock; Euroclone, Italy)in a humidified chamber with 5% CO2at 37°C. For western blot assay, the cells were cultured in a culture bottle. For immunofluorescence staining, the cells were cultured in six-well plates. The cells in the logarithmic growth phase were treated and grouped as follows: (1) Normal control group without treatment; (2) OA group: treated with OA (40 nM) for 24 hours; (3) OA + BDNF group: treated with OA (40 nM) for 24 hours, followed by adding BDNF (50 ng/mL), and incubated for 15 minutes; (4) OA + BDNF + LY294002 group:treated with OA (40 nM) for 24 hours, followed by adding BDNF (50 ng/mL) and LY294002 (4 μM), and incubated for 15 minutes; (5) OA + BDNF + K252a group: treated with OA(40 nM) for 24 hours, followed by adding BDNF (50 ng/mL)and K252a (4 μM), and incubated for 15 minutes.
Morris water maze test
On day 14 after modeling, spatial learning and memory of the rats was assessed using a Morris water maze test according to a previous study with some modifications (Bromley-Brits et al., 2011). The experimental apparatus (RWD Life Science, Shenzhen, China) was a circular water pool(diameter 150 cm; height 60 cm; containing water at 24 ± 2°C)with four equally spaced quadrants. The pool was placed in a test room containing various prominent visual cues. A translucent 10 cm × 10 cm platform, submerged 1 cm below the water surface, was hidden in the center of quadrant II during the training period and was then removed for the probe task.Memory training was performed 7 days after OA injection.The training was conducted twice every morning and afternoon, for 4 days before the probe task. Each rat was allowed to swim until it found the platform or until 120 seconds had elapsed. The rat was left on the platform for 60 seconds.During the spatial probe task, the platform was removed from the pool and the rats were allowed to swim for 120 seconds.The swim escape latency, average swim speed, time spent in the target quadrant, and number of times the animal crossed the previous location of the platform were recorded by a video tracking system (SMART, Panlab SL, Barcelona, Spain).
Western blot assay
On day 18 after modeling, the hippocampal tissues and the SH-SY5Y cells were used in the western blot assay. At the end of the final behavioral test, the rats were intraperitoneally deeply anesthetized with 10% chloral hydrate 300 mg/kg under non-stress conditions and sacrificed by rapid decapitation. The brains were quickly removed and both sides of hippocampal tissues were carefully dissected on ice. To extract the protein, tissues were homogenized in a precooled RIPA buffer (50 mM Tris-HCl, 50 mM NaCl, 5 mM ethylenediaminetetraacetic acid, 10 mM ethylene glycol tetraacetate, 2 mM sodium pyrophosphate, 4 mM paranitrophenylphosphate, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonylfluoride, 2 μg/mL aprotinin, 2 μg/mL leupeptin, and 2 μg/mL pepstatin, pH 7.5). The homogenates were incubated on ice for 30 minutes and centrifuged at 12,000 × g for 15 minutes at 4°C.
The cells for western blot assay were harvested by scraping,washed in PBS, resuspended, then homogenized and sonicated in RIPA buffer (1× PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulphate, 1 mM sodium orthovanadate, 10 mg/mL aprotinin, and 100 mg/mL phenylmethyl sulphonyl fluoride. Lysates were then centrifuged at 12,000 × g for 10 minutes at 4°C, and supernatants were collected for analysis.
The protein contents in the supernatants were determined by Bradford reagent assay (Ku et al., 2013). Protein from the hippocampal and the treated cells was mixed with loading buffer, boiled for 5 minutes, separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and then transferred to an Immobilon-P polyvinylidene difluoride(Millipore Corp.) membrane. Membranes were blocked with 5% dry milk in PBS. Proteins were probed with specific antibodies Tau (Ser199/202) (1:500; rabbit, Millipore Corp.), pTau (Ser199/202) (1:500; rabbit, Millipore Corp.),phospho-glycogen synthase kinase (pGSK)-3β (1:1000; rabbit, Cell Signaling Technology, Danvers, MA, USA), phospho-AKT (pAKT) (1:1000; rabbit, Santa Cruz Biotechnology), PP2A (1:1000; goat, Santa Cruz Biotechnology), PP2Ac-Yp307 (1:500; goat, Santa Cruz Biotechnology), Tau-5 (1:1000;mouse, Santa Cruz Biotechnology), p-tau (Thr231) (1:1000;mouse, Santa Cruz Biotechnology, p-tau (Ser396/404) (1:1000;goat, Santa Cruz Biotechnology, and β-actin (1:5000; mouse,Sigma, Santa Clara, CA, USA), and incubated overnight at 4°C.The next day, the membranes were washed four times with 0.1% Tween-20 TBS (pH 7.6) and incubated with horseradish peroxidase-conjugated anti-rabbit, anti-goat, or anti-mouse secondary antibodies (1:2000; Beyotime, Haimen, China) at room temperature for 2 hours. An enhanced chemiluminescence kit (Millipore Corp.) was used to detect immunoreactive protein bands. Protein levels were normalized to β-actin. Relative optical density of protein bands was measured following subtraction of the film background using Scion Image software (Scion Corporation, Frederick, MD, USA).
Quantitative real-time polymerase chain reaction
The fresh hippocampal tissues were put in commercial RNA extraction reagent (Roche Applied Science, Indianapolis,IN, USA) for quantitative polymerase chain reaction (qPCR)analysis. First-strand cDNA was synthesized with the use of 1 μg of total RNA (Transcriptor First Strand cDNA Synthesis Kit, Roche Applied Science). The qPCR was performed with Applied Biosystems Step One (Applied Biosystems,Foster City, CA, USA) using SYBR Green Master solution(Roche Applied Science) to measure thefluorescence intensity of amplified products. Reactions were as follows: 55°C for 2 minutes, 95°C for 10 minutes, and then 40 cycles of 95°C for 15 seconds followed by 60°C for 1 minute. Data were analyzed using the 2−ΔΔCtmethod, as previously described (Avnet et al., 2017), with β-actin as a housekeeping gene. Fold change of all groups was normalized to those of the control group. The sequences of primers are shown in Table 1. All the sequence specificities of the primers used in the current study have been verified by Primer-BLAST(http://www.ncbi.nlm.nih.gov/tools/).
Immunofluorescence staining
SH-SY5Y cells were cultured in the six-well plates. During the logarithmic phase, the cells were received different treatments. After beingfixed with 4% paraformaldehyde for 1 hour, cells were perforated with 0.5% Triton for 15 min-utes, blocked with TBS (pH 7.5) at room temperature for 30 minutes, and then incubated with either anti-PP2Ac-Yp307(1:100; goat, Santacruz Biotechnology, Santa Cruz, CA,USA) or anti-pTau (Ser199/202) (1:100; rabbit, Chemicon,Bilka, MA, USA) at 37°C for 2 hours. After washing twice with PBS for 10 minutes, the Cy3-labeled goat anti-rabbit IgG (1:200; Beyotime, Haimen, China) or FITC-labeled rabbit anti-goat IgG (1:200; Heowns, Tianjin, China) was added and incubated at 37°C for 30 minutes. Finally, after four washes with TBS for 5 minutes, the cells were sealed with quenching agent and examined under afluorescence microscope (Olympus, Tokyo, Japan).

Table 1 Oligonucleotide primer sets for real-time polymerase chain reaction
Statistical analysis
Statistical analysis was performed using SPSS for Windows(Ver. 14.0; SPSS Inc., Chicago, IL, USA). Experiments was repeated three times. Data are shown as the mean ± SD.One-way analysis of variance followed by Bonferroni confi dence interval adjustment tests was conducted to evaluate dynamic changes in all experimental data. P < 0.05 was considered statistically significant.
Results
Hippocampal injection of BDNF attenuates OA-induced cognitive decline
The Morris water maze test was performed to assess the protective effect of OA on spatial learning ability of OA-treated rats. Results of navigation paths indicated that spatial learning capacity of OA-treated rats was remarkably compared with the sham and normal control groups. The escape latencies were longer in the OA-treated rats than those in sham group(P < 0.001). In probe trials, OA-treated rats showed a fewer number of platform crosses than the sham group (P < 0.001),which indicates that OA administration impaired memory and spatial learning ability of the rats. However, administration of BDNF remarkably improved spatial learning ability in rats. BDNF-treated groups showed significantly shorter escape latencies and more platform crosses compared with the OA group (P < 0.001). Nevertheless, when K252a was added, the effect of BDNF was blocked; the escape latencies were longer and the number of platform crossings was lower than those of the OA + BDNF group (P < 0.001 and P < 0.01,respectively). When LY294002, the specific inhibitor of PI3K was added, there was no notable difference in escape latencies or the number of platform crossings compared with the OA +BDNF group (P > 0.05; Figure 1).
Exogenous BDNF reverses the cognitive dysfunction of AD rats by increasing the BDNF/TrkB/SYN mRNA levels
Normal BDNF levels and synaptic function are important for learning (Caroni et al., 2014; Petzold et al., 2015). Our qPCR results showed that compared with the sham group,the mRNA levels of BDNF, TrkB, and SYN, were significantly decreased in the OA group (all P < 0.001) and were up-regulated by administration of BDNF (all P < 0.001).However, both LY294002 and K252a inhibited the effect of BDNF on BDNF, TrkB, and SYN mRNA expressions (all P< 0.001; Figure 2). These results revealed that the exogenous BDNF may ameliorate cognitive function in AD rats by increasing BDNF levels and promoting synaptic function.
Exogenous BDNF rescues the phosphorylation of PP2A and tau protein by OA in vivo
To test the protective effect of BDNF on the over-phosphorylation of PP2A and tau protein induced by OA, we examined the levels of PP2A, PP2Ac-Yp307, p-tau (Ser396/404),and p-tau (Thr231) using a western blot assay after the stereotaxic injection. BDNF treatment inhibited the OA-induced decrease in PP2A expression and the increase in PP2Ac-Yp307, p-tau (Ser396/404), and p-tau (Thr231) levels compared with the sham group. LY294002 and K252a treatment obviously attenuated the beneficial effect of BDNF, as compared with the OA + BDNF group (P < 0.001; Figure 3).
Exogenous BDNF reduces phosphorylation of tau by enhancing the activity of PP2A in vitro
Immunofluorescence staining results showed that In the normal control group, there were hardly any PP2Ac-Yp307-positive nd pTau (Ser199/202)-positive cells; after OA treatment, the number of both PP2Ac-Yp307-positive and pTau (Ser199/202)-positive cells was obviously increased,and the staining of pTau (Ser199/202) and PP2Ac-Yp307 proteins was highly co-localized. The addition of exogenous BDNF markedly reduced positive staining of both PP2Ac-Yp307 and pTau (Ser199/202); however, when LY294002 was present, the effect of BDNF was weakened, as evidenced by increased positive staining of PP2Ac-Yp307 and pTau(Ser199/202) (Figure 4).
BDNF activates PP2A and inhibits tau phosphorylation through the PI3K/GSK-3β pathway in vitro
To further study how BDNF regulated PP2A and tau phosphorylation, western blot assay was performed to examine the associated proteins. In the OA group, PP2A expression was significantly lower (P < 0.05 and P < 0.001; Figure 5A)and the level of PP2Ac-Yp307 was markedly higher (P <0.01 and P < 0.001; Figure 5B) than the normal control and BDNF groups, but the level of tau (Ser199/202) (Figure 5C)was lower in the OA group, and the level of ptau (Ser199/202)(Figure 5D) was higher in the OA-treated group (P < 0.05 and P < 0.001) compared with the normal control and BDNF groups. No between-group differences were found in total tau protein expression (Figure 5E). In the LY294002 group, PP2A and Tau (Ser199/202) levels were significantly reduced (all P < 0.001l), and the levels of PP2Ac-Yp307 and pTau (Ser199/202) were obviously enhanced compared with the BDNF group (Figure 5A–D) (for PP2Ac-Yp307, P <0.01; for pTau (Ser199/202), P < 0.001). Compared with the normal control and BDNF groups, the OA group showed a higher level of pGSK-3β (all P < 0.001; Figure 5F), and a lower level of pAKT (all P < 0.001; Figure 5G).
Discussion
In this study, OA was used to induce the phosphorylation of PP2A and tau protein in rats and human SH-SY5Y cells.OA caused cognitive dysfunction in rats, which we suggest was the result of significantly decreased PP2A activity, an increase in tau phosphorylation, and subsequent synapse dysfunction. Exogenous BDNF reversed the cognitive dysfunction in AD rats by increasing PP2A activity and then decreasing tau phosphorylation, and also by reversing synapse dysfunction. The protective effect of BDNF on the AD brain was achieved through the PI3K/GSK-3β/AKT pathway. Our results indicate that BDNF may be a potential therapeutic target for AD.
In this study, we found that OA treatment decreased PP2A activity and dramatically increased tau protein phosphorylation both in rats and SH-SY5Y cell models of AD.Expression of the inactive PP2A was highly correlated with phosphorylated tau in SH-SY5Y cell models of AD. Tau protein is known to be involved in dementia-associated disorders, including AD, progressive supranuclear palsy,chronic traumatic encephalopathy, Pick’s disease, and corticobasal degeneration (Arendt et al., 2016). Tau protein is also known to play crucial roles in axonal transport and microtubule stabilization (Kneynsberg et al., 2017). Hyperphosphorylation of tau protein causes disruption of axonal transport, which results in an imbalance of neurotrophic factors and neuronal death (Baird and Bennett, 2013; Le et al., 2016). PP2A contributes to the regulation of tau phosphorylation in the brain, and determines an estimated 70%of tau phosphatase activity (Liu et al., 2005; Martin et al.,2013a; Martin et al., 2013b; Sontag and Sontag, 2014). PP2A mRNA and protein expression in the AD brain is confirmed to be below normal levels (Vogelsberg-Ragaglia et al., 2001;Sontag et al., 2004), while the expression of its inhibitor 2(I2PP2A) is increased (Tanimukai et al., 2005). Our results were consistent with these reports, so in vivo and in vitro AD models were successfully established.
Moreover, the present study found that OA injection into the CA3 area of hippocampus obviously accelerated tau protein phosphorylation, and markedly suppressed the expression of SYN. Emerging data have suggested that tau plays a critical role in synaptic plasticity via binding related partners (Ittner et al., 2010; Morris et al., 2011; Nisbet et al., 2015; Regan et al., 2016). Synapses form the structural basis for memory formation. Indeed, synaptic loss and dysfunction are important pathological features in the early AD brain (Pozueta et al., 2013). In one study, tau was revealed to be involved in early synaptic deficits before tau tangles formed and obvious neurodegenerative disorder occurred (Hoover et al., 2010). The multiple cellular events caused by tau hyperphosphorylation can eventually result in synaptic dysfunction and neurodegeneration (Morris et al., 2011), while inhibiting tau phosphorylation can block the mislocalization of endogenous tau (Miller et al., 2014).SYN is a specific marker for synaptic structure, constitutes synaptic vesicle-specific membrane channels, and is involved in vesicle transport and emission; SYN density has been found to be negatively correlated with the severity of dementia of AD (Wang et al., 2016). When the over-phosphorylated tau was dephosphorylated, SYN expression also increased, and the cognitive dysfunction was reversed.Thus, synaptic function may be affected by the phosphorylation of tau in the AD brain. Consistent with these reports,our results demonstrated that tau and SYN were involved in OA-induced AD.
BDNF is a key regulator of AD-associated cognitive disorders; it affects the survival of neurons, synaptic plasticity,and the memory function (Lu et al., 2014). BDNF participates in neuronal connectivity and neuroplasticity, which contributes to synaptic efficacy (Duman et al., 2000; Cotman and Berchtold, 2002). BDNF is also a promising drug target of AD because it protects adult neurons from injury in the hippocampus, cerebral cortex, and basal forebrain (Connor and Dragunow, 1998; Murer et al., 2001). Atasoy et al. (2017)reported that tau hyperphosphorylation reduced BDNF secretion and decreased BDNF levels in cortical neurons.Furthermore, the loss of BDNF has been found to promote neurite atrophy in patients with AD, and increasing BDNF levels may delay the course of AD (Nagahara et al., 2009;Nagahara et al., 2013). However, the underlying mechanism by which BDNF works on tau is unclear. We hypothesized that BDNF protects against cognitive decline via promoting neuronal survival and facilitating synaptic plasticity. The present results provide direct support for this; hippocampal OA injection obviously impaired spatial learning and memory abilities, which were strongly correlated with decreased BDNF levels. These results suggest that the inhibition of BDNF expression by over-phosphorylated tau impairs mnemonic processes in animals. Furthermore, the infusion of exogenous BDNF into the CA3 area of the hippocampus reversed the cognitive impairment induced by OA in rats via activation of PP2A and consequent dephosphorylation of tau protein, and a subsequent increase in the expression of SYN; all these effects were dependent on activation of the PI3K/AKT/GSK-3β pathway. Furthermore, in this study,when the PI3K inhibitor LY294002 was introduced, the dephosphorylation effect of BDNF on tau protein dramatically decreased, while the effect of BDNF on SYN expression and the improvement in learning and memory of AD rats were not affected. This suggests that other pathways are also involved in the effect of BDNF in the AD brain.
This study has some limitations that should be noted.First, we were not able to fully elucidate the detailed mechanisms of dephosphorylation of tau protein by BDNF treatment. We will investigate this in future experiments. Second,the in vitro experiments were performed on only one cell line, so the results should be verified using more cell lines in future work.
Taken together, this study found that BDNF expression was suppressed in the hippocampus of OA-treated rats, which resulted in obvious cognitive deficits. Intra-hippocampal injection of BDNF attenuated OA-induced cognitive impairment. Finally, the PI3K/GSK-3β/AKT pathway was involved in the mechanism of BDNF in regulating cognitive function.Our results offer a theoretical basis for BDNF as a therapeutic target for treating AD-related cognitive impairment.
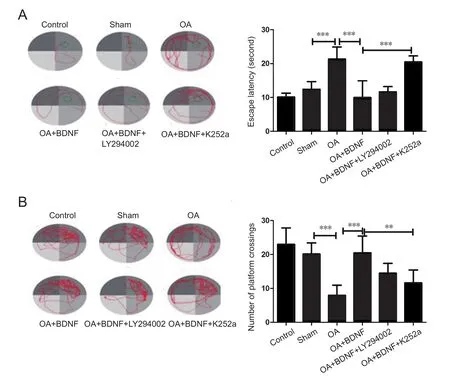
Figure 1 Exogenous BDNF improves cognitive function in a rat model of Alzheimer’s disease induced by OA.

Figure 2 Exogenous BDNF increases BDNF/TrkB/SYN mRNA levels of Alzheimer’s disease rats.
Author contributions:Drafting and critically revising the paper and agreeing to be accountable for all aspects of the work: AHX, YY, YXS, and CDZ. All authors approved thefinal version of the paper.
Conflicts of interest:There are no conflictes of interest associated with the paper.
Financial support:None.
Institutional review board statement:The study was approved by the Animal Care and Use Committee of China Medical University (ethical approval number: 1003M).
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate creditis given and the new creations are licensed under the identical terms.
Open peer reviewer:Gaoshang Chai, Jiangnan University, China.
Additionalfile:Open peer review report 1.
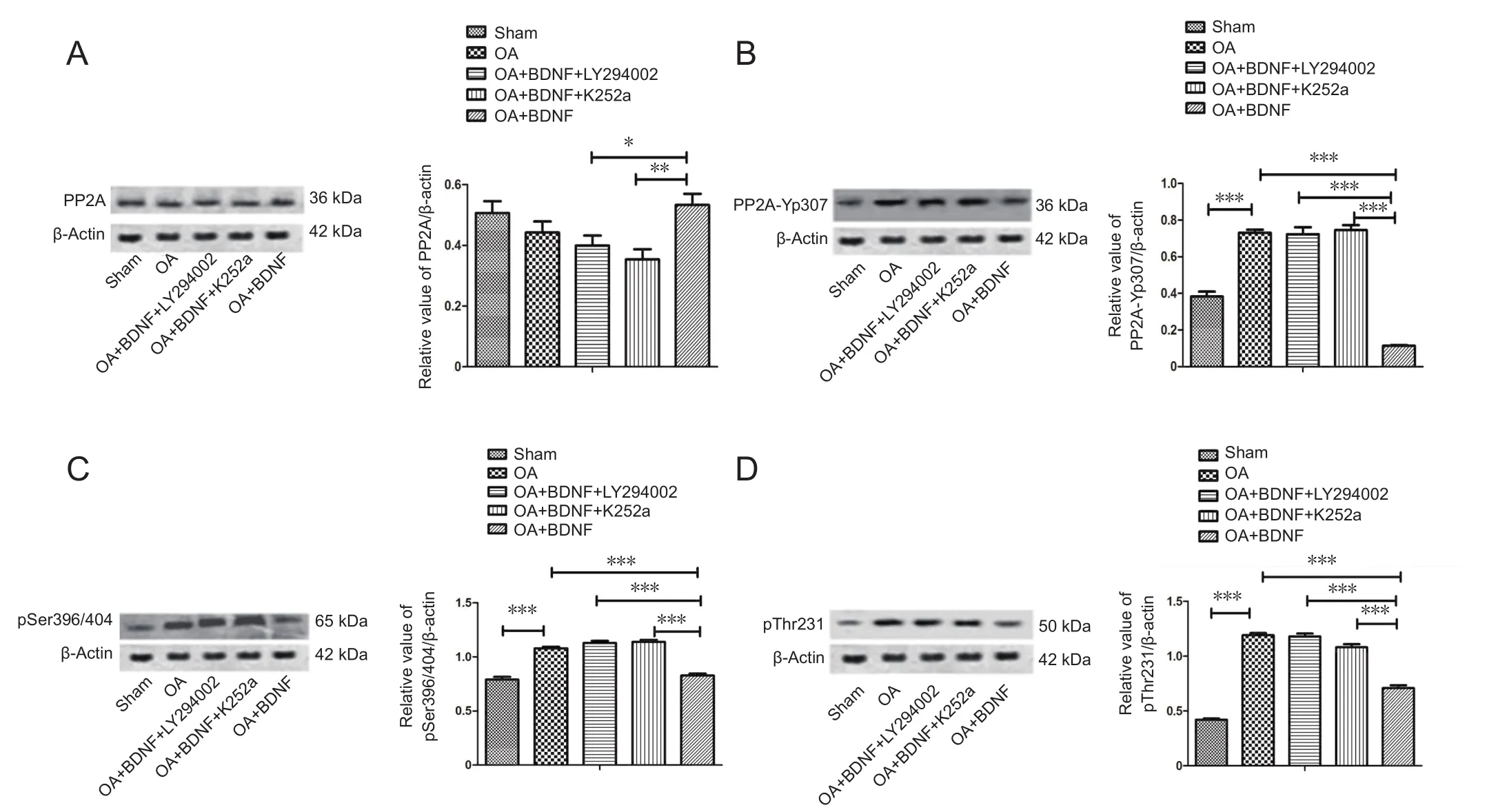
Figure 3 Exogenous BDNF rescues the OA-induced phosphorylation of PP2A and tau protein in vivo.
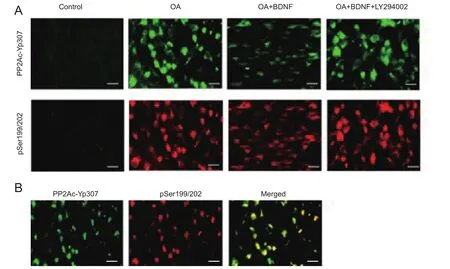
Figure 4 Correlation between phosphorylated PP2Ac (PP2Ac-Yp307) and phosphorylated tau, and the effect of exogenous BDNF in vitro.
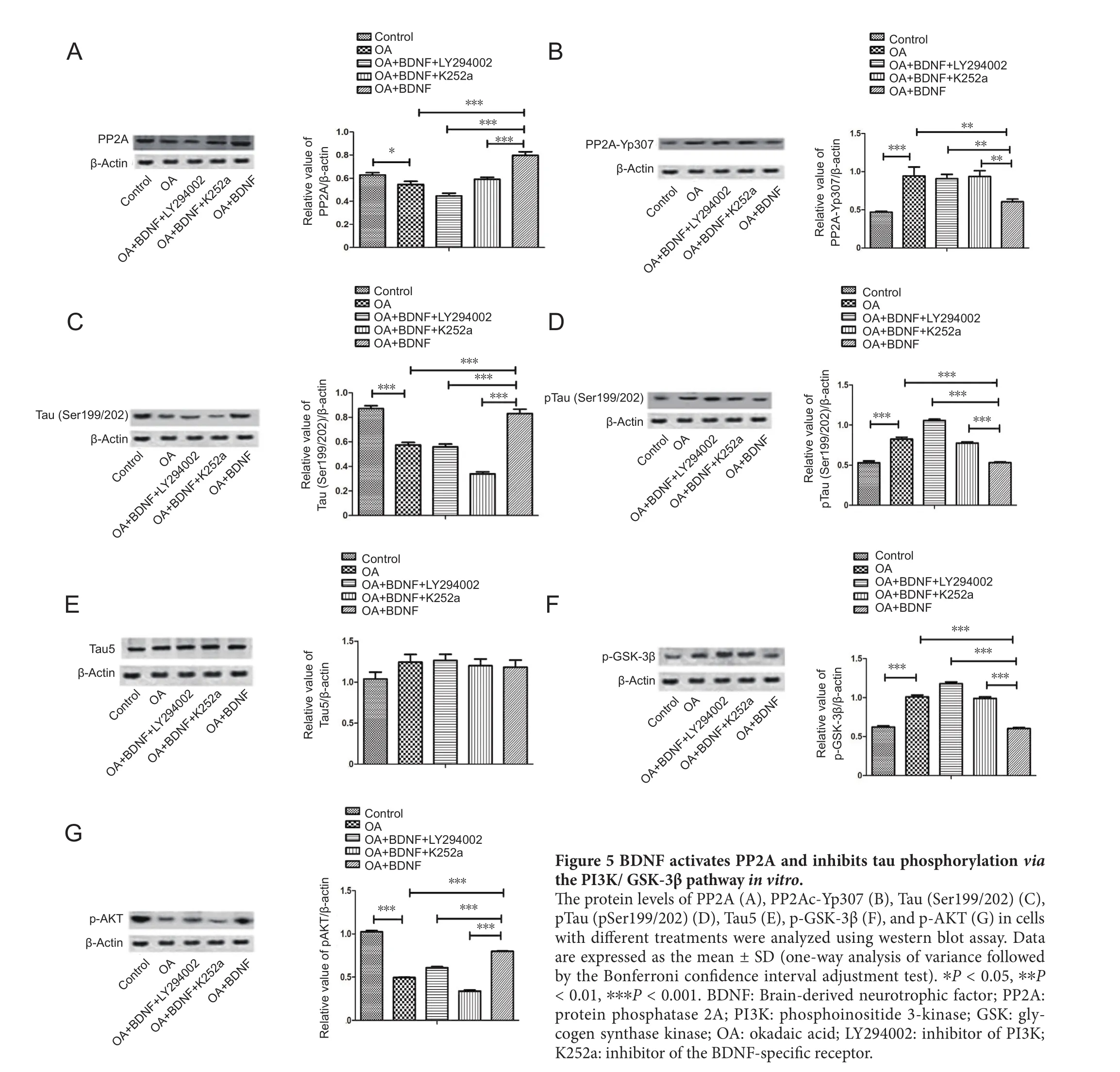
Figure 5 BDNF activates PP2A and inhibits tau phosphorylation via the PI3K/ GSK-3β pathway in vitro.
杂志排行
中国神经再生研究(英文版)的其它文章
- Huangqinflavonoid extraction for spinal cord injury in a rat model
- Lithium promotes recovery of neurological function after spinal cord injury by inducing autophagy
- Analysis of transcriptome sequencing of sciatic nerves in Sprague-Dawley rats of different ages
- Partial improvement in performance of patients with severe Alzheimer’s disease at an early stage of fornix deep brain stimulation
- Epigenetic marks are modulated by gender and time of the day in the hippocampi of adolescent rats:a preliminary study
- Dysphagia in patients with isolated pontine infarction