Consequences of redefining Alzheimer’s disease in terms of amyloid burden without regard to cognitive decline
2018-10-22StephenR.Robinson,HollyM.Brothers,MayaL.Gosztyla
Alzheimer’s disease (AD) redefined:For the past century, AD has been defined as a disease of progressive cognitive decline paired with a burden of amyloid-β (Aβ) plaques and pathologic tau tangles in the hippocampus and forebrain. However, a recent Framework paper jointly sponsored by the National Institute on Aging and the Alzheimer’s Association (Jack et al., 2018) proposes new classification guidelines for AD, which, if adopted, will have profound consequences for the future management of AD. The new guidelines redefine AD in terms of the brain’s burdens of Aβ and to a lesser extent tau, regardless of cognitive status (Figure 1). This biological approach is consistent with other diseases (e.g., type 2 diabetes) that are defined and managed in terms of biomarkers, rather than on the basis of overt symptoms. This redefinition of AD is expected to greatly facilitate progress in clinical trials and therapeutics.
Recasting Aβ burden as the primary defining feature of AD will allow the success of clinical interventions to be evaluated on the basis of whether they are able to diminish the burden of Aβ, without the need to demonstrate that cognitive decline was present or has been arrested. These relaxed guidelines will make it easier to claim success at treating AD. While the new guidelines have been proposed for sound reasons, the present authors are concerned that they will lead to adverse outcomes for patients, and could prevent progress towards a genuine cure for AD.
Advantages of a biomarker definition of AD:The influential‘Amyloid cascade hypothesis’ postulates that toxic complexes of Aβ damage the brain, eventually causing progressive cognitive decline.Over the past two decades, more than 200 clinical trials have been conducted with the specific aim of reducing the Aβ burden in AD patients order to arrest the progression of dementia. Some trials have achieved impressive reductions in Aβ burden, yet none have slowed the cognitive decline (Brothers et al., 2018).
With the worldwide number of cases of AD growing strongly,the massive size of the untapped market for AD therapeutics has led pharmaceutical companies to invest heavily in the area. However, the cost of funding a series of unsuccessful clinical trials has begun to take its toll: in January 2018, Pfizer Inc. announced that it is discontinuing its preclinical studies and Phase 1 and 2 clinical trials in AD therapeutics due to the unsustainable cost of failed clinical trials. If other pharmaceutical giants follow Pfizer’s lead, a cure for AD might be postponed indefinitely.
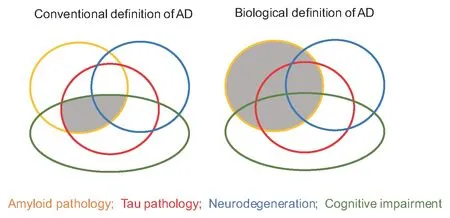
Figure 1 By making three of the conventional criteria non-essential,many more people will be classified as having AD.
Modern imaging techniques with Pittsburgh compound B positron emission tomography (PiB-PET) that enable the Aβ burden to be detected and quantified in living brains have revealed that a proportion of healthy middle-aged adults have significant Aβ burdens by midlife. A widely-held view within the pharmaceutical industry,and among proponents of the amyloid cascade hypothesis, posits that the Aβ-based therapies failed because they were conducted in late-life, whereas the pathogenic process begins much earlier(Tarawneh and Holtzman, 2009). They speculate that it should be possible to forestall the onset of AD if the Aβ deposits are targeted when they arefirst detected, in the third and fourth decades of life.By removing the dependence on cognitive decline as a defining feature of AD (Figure 1), Jack and colleagues have opened the door to the possibility of conducting clinical trials on cognitively normal people in midlife (Jack et al., 2018). Therapeutics that have already successfully lowered the brain burdens of Aβ in cognitively-impaired elders (e.g., Aducanumab, Bapineuzumab), may be repurposed to lower the Aβ burdens of middle-aged, cognitively-normal persons, thereby (definitively) treating their AD. Jack and colleagues caution that while their biological definition of AD should provide a better basis for clinical trials, the definition should not be applied as a clinical diagnosis. Nonetheless, if this approach succeeds at reducing Aβ burdens, the middle-aged may routinely begin a lifelong course of anti-Aβ therapeutics in order to stave offthe dementia that could be awaiting them in later life. There will be winners all round: peace of mind for the patient, a bonanza for big pharma, and vindication that persistent research can triumph over one of the most intractable diseases.
Ethical and clinical considerations for anti-Aβ therapeutics:Pharmaceutical companies that undertake clinical trials to attack Aβ in midlife will need to address the psychological risks associated with patients being labelled with AD, decades before cognitive decline becomes apparent. A parallel can be found in Huntington’s disease, where genetic screening is used to reveal carriers during the pre-symptomatic phase: such knowledge can enable an individual to plan for their future, yet it is also associated with higher rates of aggression, hopelessness, anxiety, depression and suicidal ideation (Anderson et al., 2016). The imperfect correlation between Aβ load and subsequent conversion to AD raises additional concerns. A considerable body of evidence has demonstrated that many elderly individuals with high levels of Aβ never develop progressive cognitive decline or dementia (Perez-Nievas et al., 2013).Initiating anti-Aβ therapies in the absence of any cognitive deficits will inevitably result in some individuals receiving unnecessary treatment, while increasing their risk of depression and anxiety.
Another consideration relates to the adverse outcomes that commonly arise from depleting the brain of Aβ. Clinical trial reports of anti-Aβ therapeutics indicate that the incidence of cerebral edema and micro-bleeds increases nearlyfive-fold following the initiation of therapy, and can lead to headache, confusion, nausea and gait disturbances. Indeed, these unpleasant symptoms are so prevalent, occurring in up to half of the patients in some immunotherapy trials,that they are referred to euphemistically as ‘ARIA’ (amyloid-related imaging abnormalities). Other adverse outcomes include increased rates of meningoencephalitis and re-emergent infections (Brothers et al., 2018). This situation begs the question of whether it is ethical to knowingly expose cognitively normal people to the risk of these adverse outcomes, particularly when it will not be known for decades whether anti-Aβ therapy in midlife reduces the risk of developing dementia in old age.
In addition to its involvement in AD, Aβ is produced by the brain throughout life where it appears to serve several important physiological roles, including intercepting pathogens that infiltrate the brain, patching breaches of the blood-brain barrier and assisting in the consolidation of memories [reviewed by Brothers et al. (2018)]. Thus, it may prove necessary to screen participants in anti-Aβ clinical trials for the remainder of their lives, to ensure that the neutralisation of Aβ’s physiological functions does not increase the rates of cerebral infection, micro-bleeds or memory loss.
Evidence suggests that Aβ may assist the nervous system to recover from injury (Brothers et al., 2018). Aβ expression increases within hours of a traumatic brain injury (TBI) in both humans and animals, including models without AD-related transgenes. A study of 18 TBI patients found that the titers of Aβ in the brain’s interstitialfluid are positively associated with cognitive status (Brody et al., 2008). Mice that have a knockout of the gene coding for β-site amyloid precursor protein cleaving enzyme 1 (BACE1) are unable to cleave Aβ from its precursor, and following a TBI they show impaired spatial memory when compared to wild-type mice.Treatment of these BACE1 knockout mice with exogenous Aβ improves their memory, while the treatment worsens memory in wild-type mice, suggesting that the levels of Aβ normally produced in response to TBI are tuned within appropriate limits (Mannix et al., 2013).
The presence of Aβ promotes recovery from other forms of injury. BACE1 knockout mice and Aβ-precursor protein (APP) knockout mice both have a 40% survival rate within 4 hours of cerebral ischemia, compared to 100% survival rates in wild-type mice (Koike et al., 2012). Likewise, in mice subjected to a spinal cord injury,prevention of Aβ production by BACE1 knockout or γ-secretase inhibition results in impaired motor recovery and more extensive white matter damage (Pajoohesh-Ganji et al., 2014). Conversely,AD-transgenic rats, which overexpress Aβ, have reduced infarct volumes compared to wild-type rats following occlusion of the middle cerebral artery; the transgenic rats perform better on tests of spatial memory and fear conditioning than the wild-type rats,despite worse outcomes on motor tasks (Clarke et al., 2007). Further, in four different mouse models of multiple sclerosis, intraperitoneal injections of hexameric Aβ40or Aβ42led to improvements in motor function, remyelination of lesions and reduced inflammation (Grant et al., 2012). Collectively, these studies suggest that focusing AD therapeutics on the depletion of Aβ may impair the capacity of patients to recover from neurological injuries, of which the elderly are at an increased risk.
Conclusion:While focusing AD drug development on the reduction of Aβ burden offers certain advantages for clinical trials, it may result in unintended consequences, including a diminished capacity to recover from stroke and brain injury, psychological repercussions of believing they have AD, as well as the risk of adverse side-effects like ARIA and meningoencephalitis. If active immunotherapies are used, the autoimmune response against Aβ will be lifelong, potentially extending these risks over decades.
What if the amyloid cascade hypothesis is wrong? Some researchers have argued for a causative role of tau in AD pathogenesis, whereas others have proposed that Aβ plaques are part of an innate response to brain injury (Robinson and Bishop, 2002). Animal and human studies have shown that Aβ deposition can be induced by a wide range of events that are injurious to the brain including TBI, stroke and hypoxia (Brothers et al., 2018). Although the Framework paper acknowledges that there is uncertainty regarding whether Aβ plaques are the cause or a consequence of the disease process (Jack et al., 2018), the biological definition implies that the presence of a higher-than-normal burden of Aβ is AD. In effect,the amyloid cascade has been elevated from a hypothesis to a theorem, with the support of the National Institute on Aging and the Alzheimer’s Association. This shift in status is bound to discourage research into alternative therapeutic targets. If Aβ deposition is a consequence of AD rather than its cause, then it could take 20 to 30 years before we learn that anti-Aβ treatments administered continuously from midlife do not prevent cognitive decline in late life.In the meantime, the impetus for developing alternative treatments will have been lost.
Ironically, we may never know that anti-Aβ treatments are ineffective at preventing AD, because progressive cognitive decline in the absence of an Aβ burden does not meet the diagnostic de finition of AD. For this reason, and those outlined in the preceding paragraphs, we caution against adopting the proposed biological definition of AD in research or in clinical trials.
Stephen R. Robinson*, Holly M. Brothers, Maya L. Gosztyla
Discipline of Psychology, School of Health and Biomedical Sciences, RMIT University, Bundoora, Victoria, Australia; Institute for Breathing and Sleep, Austin Health, Heidelberg, Victoria,Australia (Robinson SR)
Department of Psychology, The Ohio State University, Columbus,OH, USA (Brothers HM)
Department of Neuroscience, The Ohio State University,Columbus, OH, USA (Gosztyla ML)
*Correspondence to:Stephen R. Robinson, PhD,stephen.robinson@rmit.edu.au.
orcid:0000-0002-0987-0075 (Stephen R. Robinson)
Received:2018-05-24
Accepted:2018-08-07
doi:10.4103/1673-5374.241456
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
杂志排行
中国神经再生研究(英文版)的其它文章
- Huangqinflavonoid extraction for spinal cord injury in a rat model
- Lithium promotes recovery of neurological function after spinal cord injury by inducing autophagy
- Analysis of transcriptome sequencing of sciatic nerves in Sprague-Dawley rats of different ages
- Exogenous brain-derived neurotrophic factor attenuates cognitive impairment induced by okadaic acid in a rat model of Alzheimer’s disease
- Partial improvement in performance of patients with severe Alzheimer’s disease at an early stage of fornix deep brain stimulation
- Epigenetic marks are modulated by gender and time of the day in the hippocampi of adolescent rats:a preliminary study