血尿酸与非酒精性脂肪肝的相关性研究进展
2018-10-11王丽莹李强
王丽莹,李强
非酒精性脂肪肝(NAFLD)病理学定义为肝脏脂肪含量超过肝总重量的5%,要求男性饮用乙醇量<20 g/d(140 g/周),女性饮用乙醇量<10 g/d(70 g/周)[1]。在西方国家,NAFLD患病率为20%~50%,亚洲为5%~18%[2]。并且NAFLD在各个年龄人群均有发病,在非肥胖型成年人中患病率为10%~15%,肥胖型成年人中为70%,特别值得注意的是在10岁以下儿童中其患病率约为10%,青少年中为17%,肥胖儿童甚至增长至40%~70%[3]。NAFLD患病率也在逐年增加,LU等[4]于我国浙江进行的历时8年的随访研究表明,NAFLD患病率为35.47%,新发患者为17.30%,患病率增长46.46%,仅有6.31%患者恢复正常。血尿酸(SUA)是脊椎动物体内嘌呤代谢的终产物,其中人类由于嘌呤代谢过程短而迅速,与其他动物相比其SUA水平较高,较易发生高尿酸血症。而SUA水平增加促进胰岛素抵抗(IR)、线粒体和内质网氧化应激、NLRP3炎症复合体的发生发展。SUA不仅与痛风、关节炎及肾脏疾病有关,还与包括心血管疾病和代谢综合征相关的一系列疾病密切相关,且SUA水平升高在2型糖尿病(T2DM)、NAFLD发生发展中起至关重要的作用。
NAFLD具有广泛的组织学图谱,包括简单脂肪变性、脂肪性肝炎、肝纤维化和肝硬化。简单脂肪变性被认为是一个良性的过程,被称为“沉默的肝脏疾病”,CHALASANI等[5]研究表明,1%~5%的简单脂肪变性将进展为肝硬化,但脂肪性肝炎进展为肝纤维化及肝硬化的比例约为30%,其甚至可进展为终末期肝病和肝癌。CLEMENTE等[3]和WONG等[6]预测,在下一个10年NAFLD将成为肝脏移植的最重要病因,甚至发生在少年时期。可见加强NAFLD的综合管理意义重大。为此,本文结合SUA病理生理作用与NAFLD流行病学特点、机制等,阐述SUA与NAFLD的相关性及相关机制,为有效管理与监测NAFLD提供理论支持。
1 SUA的病理生理作用
SUA生成及代谢途径如图1[7]。人体内SUA的来源包括内源性SUA(来自细胞代谢的核蛋白)和外源性SUA(饮食),而其排泄受肾脏血浆流量、肾小球滤过率等影响,生成和排泄的动态平衡是SUA稳定的前提。因此产生过量或排泄减少均会引起SUA水平增加。其常见的原因包括代谢综合征(MS)、高糖高脂饮食、肾功能障碍等。SUA不仅与痛风、关节炎及肾脏疾病有关,还与心血管代谢疾病密切相关,包括心血管疾病和与MS相关的一系列疾病[8]。SUA在体内既有抗氧化作用,又有促氧化作用。
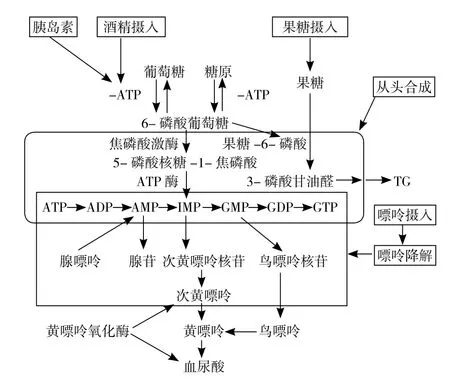
图1 SUA生成及代谢途径Figure 1 Production and metabolic pathway of SUA
1.1 SUA的抗氧化作用 SUA可以在尿酸酶的氧化作用下转化为尿囊酸,在进化过程中,尿酸酶基因发生沉默,人体无法分解SUA,所以与其他哺乳动物相比其SUA水平较高,较易发生高尿酸血症。但是,这种相对高SUA状态对于人体仍具有保护作用,体内SUA可以通过中和氧化剂分子来起到抗氧化的作用;常见的氧化剂分子有羟自由基、过氧化氢和过氧亚硝酸盐等[9]。KAMOGAWA等[10]采用电子自旋共振(ESR)法评估SUA、谷胱甘肽、依达拉奉清除氧自由基的能力,结果发现,对超氧阴离子自由基的清除速率常数最高的是SUA,并且SUA对甲基自由基及叔丁基过氧自由基清除速率常数仅略低于超氧阴离子自由基。对于部分中枢神经系统退行性变和神经炎性疾病,如多发性硬化症、帕金森病和急性卒中,研究发现迅速提高SUA水平有助于缓解上述疾病的病情并延缓疾病进展[11],但值得注意的是慢性SUA水平升高会加重卒中风险。ASHTARI等[11]的Meta分析共纳入研究对象2 493名,其中帕金森病患者1 217名,健康对照者1 276名(性别、年龄匹配),应用Stata 12.0统计数据软件进行统计分析,结果显示,帕金森病患者SUA水平明显低于健康对照者,且男性患者较女性患者差异更明显。不仅在神经系统,在心血管系统SUA也具有明显的抗氧化作用,体外培养的主动脉内皮细胞及脂肪细胞发现,SUA可以阻断神经元中过氧亚硝基阴离子介导的亚硝基化作用,减少白细胞渗出,但其对过氧亚硝基阴离子无直接的清除作用,而是通过减少内皮细胞对一氧化碳的利用度,进而抑制一氧化碳参与的亚硝基化作用[12]。在人类进化的过程中,SUA的抗氧化作用为生存优势之一,这对人类的生存发展意义重大。
1.2 SUA的促氧化作用 SUA的抗氧化作用具有局限性,疏水环境对SUA的抗氧化作用是不利的,即SUA只有在亲水环境中表现出显著的清除氧自由基的抗氧化作用,但在亲脂的环境下由于其不能打破脂质膜而失去抗氧化作用。氧化脂质可以使SUA转变为促氧剂,SUA与其他氧化剂反应也可以转变为促氧剂,进而产生氧自由基,而且产生的氧自由基靶目标为脂质〔低密度脂蛋白(LDL)、脂质膜〕。与未分化脂肪细胞相比,分化脂肪细胞的表型特征是具有较高的尿酸盐摄取率、尿酸盐转运蛋白的超表达并且有大量活性氧(ROS)产生。有研究应用硝基氮蓝四唑(NBT)测量ROS产生量,与无SUA组相比,SUA水平为1~15 mg/dl时,脂肪细胞分化过程中产生了大量ROS,随着SUA水平及培养时间的增加,ROS产生量明显增加,但未分化脂肪细胞组无明显变化;应用ROS特异性荧光探针H2DCFDA检测ROS,也发现相同结果,可见SUA可以诱导分化脂肪细胞产生ROS;应用尿酸盐跨膜转运抑制剂丙磺舒和苯溴马隆处理细胞,发现两种抑制剂均可阻止ROS产生,可见SUA必须进入细胞才能诱导ROS产生[13]。研究发现,SUA的这种作用可以被细胞渗透超氧化物歧化酶MnTMPyP阻断,也可以被还原型烟酰胺腺嘌呤二核苷酸磷酸(NADPH)氧化酶的抑制剂〔N-乙酰半胱氨酸、夹竹桃麻素、二苯基碘(DPI)〕削弱,但线粒体呼吸链阻滞剂〔2-噻吩甲酰三氟丙酮(TTFA)、鱼藤酮〕并未影响SUA的作用结果,表明SUA诱导ROS产生取决于NADPH氧化酶而不是线粒体呼吸链[14]。以上研究证明,SUA可以通过参与细胞内NADPH氧化酶过氧化物生成系统成为助氧化剂。SUA的促氧化作用与NAFLD、MS等多种代谢性疾病密切相关[7]。
2 SUA与NAFLD关系及相关机制
2.1 SUA与NAFLD的相关性 随着研究的深入,越来越多的证据表明SUA与NAFLD的发生发展密切相关,但二者的因果关系,仍存在争议[15-22]。LIU等[15]进行的横断面研究共纳入1 365名肥胖成年人,通过超声诊断NAFLD,NAFLD发生率男性为71.5%,女性为53.8%;NAFLD组SUA水平和高尿酸血症发生率均显著高于对照组;去除IR、MS和其他潜在混杂因素后,发现升高的SUA与NAFLD发生率的增加独立相关,调整后的OR值为1.528~2.031(P<0.001);使用多变量分数多项式(MFP)建模,模型显示,SUA水平与NAFLD的发生风险独立相关且有线性相关关系;结构方程模型(SEM)显示,SUA可能直接增加NAFLD的发生风险并且增加空腹胰岛素、血压、三酰甘油(TG)、高密度脂蛋白胆固醇(HDL-C)水平。除了简单的脂肪定性研究,LIN等[16]进行了肝脏脂肪定量(LFC)研究,结果显示,LFC为SUA的独立影响因素,二者呈正相关,LFC>10%组较LFC<5%组SUA水平明显升高。LIU等[17]进行的Meta分析共纳入了9篇高质量的前瞻性研究,其中7篇是基于SUA与MS相关性的研究(共入选23 081名男性及12 195名女性),有6篇提供原始数据并且通过重新提取分析(共包括34 222名参与者,其中5 032名MS患者),结果显示,SUA水平每增加1 mg/dl,MS发生率男性增加5%,女性增加9%,而且在同等SUA水平上年龄<52岁的女性与同年龄男性及老年女性相比更容易发展为MS;另2篇是基于SUA与NAFLD相关性的研究(入选4 492名男性及8 139名女性),结果显示,SUA水平每增加1 mg/dl,NAFLD发生风险约增加1.03。为了排除性别对研究结果的影响,YU等[18]进行了一项性别特异性纵向研究,包括14 442名健康参与者(8 715名男性和5 727名女性),用Cox比例风险模型来量化SUA水平与MS的关联性,经过6年的随访,有4 215名参与者(2 974名男性,1 241名女性)新发以NAFLD为主的代谢性疾病,并且SUA每增加1 mg/dl,风险比男性为1.094、女性为1.148。国内外多项研究也相继验证以上观点[16,18-21]。
然而,流行病学研究无法从分子机制方面阐明SUA水平升高是否为NAFLD发生发展的决定性因素。WAN等[22]通过体内实验及体外实验证明了SUA在NAFLD发生发展中的核心作用;体内实验选取C57BL/6小鼠,随机分为高UA组(HUA组)、高脂饮食组(HFD组)及正常饮食组(SCD组),经过8周喂养后,分析小鼠血清学指标及肝内TG水平的组间差异,其中肝内TG水平通过HE染色和油红O染色测定,结果显示,HUA组及HFD组小鼠与SCD组相比SUA、血清TG水平和肝内TG水平均显著升高,而且与HFD组相比,HUA组肝内TG水平更高;体外实验选取人肝癌细胞系HepG2和人正常肝脏细胞系LO2,根据培养液不同分为SUA组、SUA+游离脂肪酸组、SUA+丙磺舒组,每个实验组又根据SUA水平的不同分为4个亚组,培养48 h后,通过油红O染色比较细胞内TG水平,结果显示,细胞内TG水平随着SUA水平升高而增加,SUA增加了游离脂肪酸诱导细胞内TG的积累,由于丙磺舒可以阻断SUA进入细胞,SUA+丙磺舒组细胞内TG水平明显低于其他两组,SUA可以诱导HepG2细胞内的TG积累,同时丙磺舒显著削弱SUA的这种促进作用。可见无论体内实验或体外实验均证明,SUA可增加肝内TG沉积,在NAFLD发生发展中起关键作用。
2.2 SUA与NAFLD发生的相关机制 SUA与NAFLD发生的可能机制可以概括为图2[23],其包括MS、肥胖、高果糖饮食等导致SUA水平增加,引起胰岛素敏感性降低、线粒体氧化应激、NLRP3炎症复合体激活,进而导致体内ROS增加及内质网应激,最终导致NAFLD,而且在NAFLD发生过程中各种危险因素又相互促进,是一个复杂的过程。
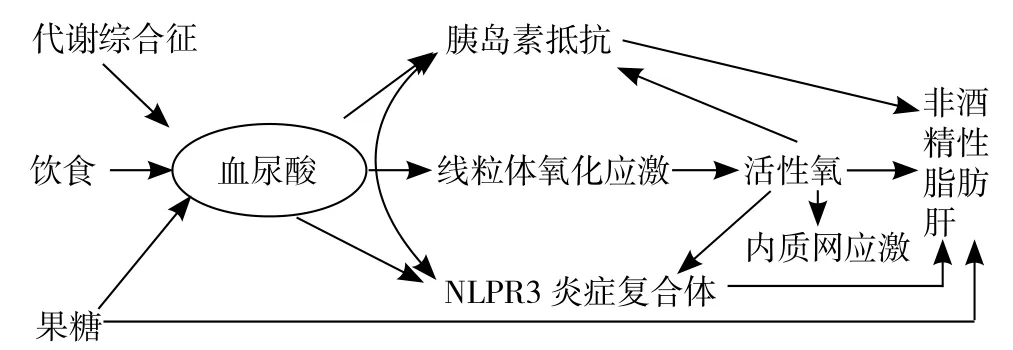
图2 高尿酸血症与NAFLD发生相关机制Figure 2 Relationship between hyperuricemia and NAFLD
2.2.1 高尿酸血症与IR 胰岛素的生理作用之一是其可以作用于近端肾小管,增加SUA的重吸收,导致体内SUA水平增加,但LI等[24]研究发现,SUA水平增加可以通过降低体内一氧化氮(NO)的生物利用度进而引起IR。而且胰岛素是细胞色素P450 4A(CYP4A)的主要抑制剂,因此IR显著增加了ROS,促进氧化应激。胰岛素的主要抑制作用是细胞毒性和脂质过氧化。这些细胞毒性物质及脂质过氧化产物可以扩散到细胞外,影响枯氏细胞和肝星状细胞诱导的核转录因子κB(NF-κB)通路,导致肿瘤坏死因子α(TNF-α)等促炎因子增加[25]。ZHI等[26]进行的小鼠实验模型探究高尿酸血症引起心肌细胞IR的具体机制,研究将小鼠心肌细胞暴露于高SUA的环境中,用荧光的方法量化葡萄糖的吸收,探究IR程度和ROS产生情况,通过免疫印迹技术检测胰岛素受体水平,研究发现,高SUA可以抑制胰岛素诱导的葡萄糖吸收,通过抑制蛋白激酶B的磷酸化和增加肝脏、肌肉、脂肪组织胰岛素受体底物磷酸化,进而抑制胰岛素信号传导,降低胰岛素的敏感性,增加IR程度。高SUA也可诱导氧化应激和激活NLRP3炎症复合体,进而加重IR[27]。相关前瞻性研究也进一步验证,药物降低SUA水平可以增加胰岛素的敏感性,该研究共纳入肾功能正常门诊患者121例,包括高尿酸血症组(73例)和SUA正常组(48例),将高尿酸血症组随机分为实验组(40例,应用别嘌呤醇)和对照组(33例,应用安慰剂),经过3个月的观察,结果显示,实验组空腹血糖、空腹胰岛素、稳态胰岛素评价指数(HOMA-IR)较基线水平均明显改善,但其他两组没有明显差别[28]。可见降低SUA水平有助于增加胰岛素敏感性。
2.2.2 高尿酸血症与线粒体、内质网氧化应激 在肝脏脂肪变性的过程中,线粒体、内质网氧化应激起至关重要的作用。线粒体氧化应激抑制三羧酸循环中顺乌头酸酶活性,柠檬酸堆积,从而促进肝细胞中的脂肪沉积与合成。LANASPA等[29]提出高SUA会激发细胞线粒体氧化应激,诱导肝细胞中脂质重新合成,最终使细胞内脂质合成增加;该研究通过测定DCF荧光强度比较氧化应激情况,结果显示,随着SUA水平的增加,DCF荧光强度明显增加,而且别嘌呤醇能显著降低高SUA组DCF荧光强度;研究人员应用线粒体染色(Mitsox)发现,与对照组相比,高SUA组线粒体荧光复合物显著增加,通过JC-1染色测定线粒体膜电位也发现SUA可明显降低线粒体膜电位;可见SUA诱导细胞氧化应激主要发生在线粒体。为探究SUA促进线粒体氧化应激是否与NADPH氧化酶亚基NOX4易位到线粒体有关,将HepG2细胞暴露于高SUA环境培养24 h,应用共聚焦显微镜及ACAA2标记线粒体,研究发现NOX4位于正常HepG2细胞的细胞质中,当细胞暴露于SUA中24 h后,NOX4转移到线粒体中,应用免疫组化法分离出的NOX4水平也随SUA水平的增加明显增加;相反,NOX4基因敲除细胞与对照组相比,线粒体荧光复合物水平显著下降;可见SUA联合诱导线粒体氧化应激与NADPH氧化酶亚基NOX4的线粒体易位有关。
氧化应激产生的ROS也会引起内质网氧化应激,导致肝脏脂肪沉积。内质网是调节蛋白质合成后折叠及脂质类固醇产生的场所,任何原因引起内质网生理功能紊乱均会引起内质网对蓄积在网腔内的错误折叠或未折叠蛋白质进行处理,进而维持细胞的正常功能。当内质网应激时,会激活未折叠蛋白效应(UPR),以保持内质网稳态,但当UPR不足时,将诱导细胞损伤甚至凋亡。内质网氧化应激激活固醇调节元件结合蛋白-1c(SREBP-1c),调节脂质代谢,促进脂肪沉积。SUA可以诱导编码内源性脂质酶基因的表达,进而增加内源性SREBP-1c的表达,诱导内质网氧化应激,当SUA水平>6 mg/dl时,HepG2细胞及大鼠干细胞内TG水平明显增加,SUA水平为12 mg/dl时,细胞内会发生内质网氧化应激[30]。高尿酸血症引起的线粒体、内质网氧化应激与多种代谢性疾病密切相关[31]。
2.2.3 高尿酸血症与NLRP3炎症复合体 NLRP3炎症复合体是细胞内多蛋白复合物,为固有免疫的重要组成部分,包括NOD样受体(NLR)、凋亡相关微粒蛋白(ASC)、效应分子胱冬肽酶-1前体(pro-caspase-1),能够调节caspase-1的活化进而在固有免疫防御的过程中促进细胞因子前体pro-IL-1β和pro-IL-18的切割成熟,诱导分泌白介素(IL)-1β和IL-18,在机体免疫反应和疾病的发生中具有重要作用。而且NLRP3炎症复合体在肥胖、IR、脂质代谢异常及肝细胞脂肪变性中均起到重要作用[32]。CAI等[33]通过蛋白质印迹法及PCR法检测肝脏枯否细胞中NLRP3、ASC基因、caspase-1水平,并应用酶联免疫吸附试验法测定IL-1、IL-18水平,发现在NAFLD小鼠模型中,NLRP3通过诱导枯否细胞分泌IL-1β和IL-18发挥促炎作用;相反,NLRP3基因敲除小鼠模型在相同条件下其NLRP3炎性小体的上调和促炎因子IL-1β、IL-18的激活均明显受抑制;实验证明NLRP3炎症复合体在NAFLD发生发展中起重要作用。SUA不仅促进肝细胞变性、增加胰岛素信号传导障碍,而且直接诱导肝细胞脂肪沉积和IR,还可以通过激活NLRP3炎症复合体活性促进以上过程发展。用实时荧光定量PCR法检测发现,经过高SUA喂养的小鼠与正常饮食及高脂喂养小鼠相比,肝细胞内NLRP3、caspase-1、IL-1β、IL-18 mRNA水平明显上调,而且用蛋白质印迹法及酶联免疫吸附试验法检测到NLRP3、caspase-1、IL-1β、IL-18水平均明显高于对照组,用药物降低SUA水平后NLRP3表达水平明显下降[33]。不仅在体内,用类似的方法培养HepG2和LO2细胞系,也发现高SUA培养的细胞中NLRP3、caspase-1、IL-1β、IL-18水平增加,可见无论在体内还是体外,SUA均具有诱导NLRP3炎症复合体表达的作用[34]。SUA可能是NAFLD和IR新的治疗靶点。
2.2.4 SUA与果糖代谢 与葡萄糖相比,果糖更容易诱导小鼠发生IR。果糖可以诱导大鼠MS、氧化应激、内皮功能障碍、脂肪肝、蛋白尿和肾脏疾病的发生。RONCAL-JIMENEZ等[35]的动物实验发现,与果糖相比,在同样的摄入量下葡萄糖对MS等没有明显的诱导作用。对果糖的这种特殊的诱导作用存在多种假设,目前为大家普遍接受的是果糖的特殊代谢途径,即果糖与葡萄糖的初始代谢不同,果糖代谢的第一个酶是果糖激酶(KHK),其主要发生在肝脏,是快速的、没有任何负面反馈的过程,使细胞内磷酸盐和ATP迅速下降。细胞内磷酸盐的减少刺激AMP脱氨酶(AMPD)催化降解为一磷酸肌苷,最后代谢产物为SUA,进而使细胞内SUA水平增加[36]。细胞内尿酸由肝脏释放入血引起SUA水平增加。KHK包括HKH-c和KHK-a两个亚型,其中HKH-c迅速消耗ATP并生成SUA,KHK-a缓慢消耗ATP,ISHIMOTO等[37]研究表明,HKH-c和KHK-a基因敲除的小鼠中,没有MS和脂肪肝的发生,但当选择性敲除KHK-a基因时,在摄入总热量及果糖量相同的情况下,上述疾病的发生比例明显增加。SUA还可以激活核转录因子、碳水化合物反应结合蛋白(ChREBP),增加肝细胞内KHK水平,而KHK水平增加时TG对果糖的反应也会增强,导致脂肪堆积及MS。LANASPA等[38]将HepG2细胞于不同水平SUA培养基中培养72 h,应用免疫组化法检测KHK水平,发现随着SUA水平增加,KHK水平增加并且具有剂量依赖性,而且在SUA水平为750 mmol/L时,KHK水平于暴露24 h后增加35%,在暴露48 h和72 h时具有更大的增量。为探究果糖诱导的KHK水平上调是否同样是SUA介导的,研究将HepG2细胞暴露于果糖中培养72 h,应用免疫组化法检测到KHK水平明显上调,但应用SUA KHK水平显著降低[35]。可见SUA可以诱导KHK表达,并且可以促进果糖诱导KHK表达。有研究通过免疫沉淀法检测ChREBP基因乙酰化状态来确定ChREBP基因水平,发现SUA诱导KHK表达的作用是通过ChREBP介导的[35]。
3 小结及展望
SUA的病理生理作用决定其在人体代谢中的独特性。越来越多的证据表明体内SUA水平与NAFLD发生发展密切相关[8,31,39-40]。SUA可以促进IR及线粒体、内质网氧化应激的发生发展,并且SUA可以诱导果糖代谢,这些机制均与NAFLD的发生发展有关。但目前仍缺乏大规模前瞻性研究,应进一步探究并明确SUA与NAFLD的因果关系,为有效管理相关代谢性疾病提供理论依据。
