β-甘油磷酸钠修复含铀地下水
2018-09-25丁德馨李殿鑫
黄 超,张 辉,胡 南,丁德馨*,李殿鑫,孙 静
β-甘油磷酸钠修复含铀地下水
黄 超1,2,张 辉1,2,胡 南1,2,丁德馨1,2*,李殿鑫1,2,孙 静1,2
(1.南华大学,铀矿冶生物技术国防重点学科实验室,湖南 衡阳 421001;2.南华大学,极贫铀资源绿色开发技术湖南省重点实验室,湖南 衡阳 421001)
利用微模型实验,研究β-甘油磷酸钠修复含铀地下水的效果,监测厌氧培养过程中硝酸盐、硫酸盐、磷酸盐、pH值和铀浓度的变化,分析沉积物中铀的化学形态以及U(IV)的比例.结果表明:厌氧培养25d后,β-甘油磷酸钠组和甘油组的铀浓度分别从2.96, 2.99mg/L降低到0.030, 0.044mg/L,达到了我国《铀矿冶辐射防护和环境保护规定(GB23727-2009)》规定的排放标准.β-甘油磷酸钠作为碳源和磷源能够降低沉积物中可交换态和碳酸盐结合态铀的比例,并能提高沉积物中铁锰氧化物结合态,有机结合态和残渣态铀的比例;厌氧培养25d后,β-甘油磷酸钠组沉积物中U(IV)的比例达到了91.79%,比甘油组提高了28.82%,厌氧培养45d后,β-甘油磷酸钠组沉积物中U(IV)的比例基本保持不变,而甘油组则下降了7.98%.β-甘油磷酸钠作为碳源和磷源,能够促进U(VI)的生物还原和矿化从而将铀原位固定.
β-甘油磷酸钠;含铀地下水;生物还原;生物矿化;原位固定
地下水铀污染是铀矿开采和加工领域亟待研究解决的重大问题[1-2].铀在地下水中主要以UO22+、UO2(OH)+、(UO)2CO3、UO2(CO3)22-, UO2(CO3)34-、(UO)2(OH)22+、(UO2)3(OH)5+等形态存在[3].这些形态的铀溶解度高,极易随着地下水的流动而迁移,从而造成地下水中更大范围的污染[4].由于铀的重金属毒性和放射性辐射,进入人体后主要积蓄于肾脏、肝脏和骨骼中,容易引起肝脏肾脏病变和诱发癌症[5-7].
修复铀污染地下水的技术中,原位生物修复是应用最为广泛的一种修复方法[8].与传统的物理化学方法相比,生物修复法具有效率高、成本低、不会产生二次污染物等优点[9-10].通常通过加入电子供体激活微生物发生酶促反应,将水溶性的U(VI)还原成不溶于水的U(IV),或者激活微生物发生代谢使其代谢过程中产生的SO42-、PO43-、CO32-、OH-等配位体与溶液中的铀矿化生成难溶的沉淀[11-12].目前,大多数的研究主要集中在微生物还原U(VI)[13-14].
但是有研究发现,生物还原生成的U(IV)暴露于氧气或硝酸盐之后容易被氧化[15].Mehta等[16]发现磷酸盐的存在能限制铀在水体中的迁移,且铀的磷酸盐是一种不溶于水也不容易被氧化的沉淀[17],但是如果直接添加无机磷到地下水体中,容易导致生成金属磷酸盐沉淀,局部堵塞注液井[18].Newsome等[19]发现有机磷能为微生物修复提供碳源和磷源,且能生成稳定的不易被氧化的U(IV)-磷酸盐矿物.
β-甘油磷酸钠是一种碳源和磷源,本研究采用微模型实验探索了β-甘油磷酸钠作为碳源和磷源对原位修复含铀地下水的效果,监测了微模型中硝酸盐、硫酸盐、磷酸盐、pH值和铀的浓度变化,采用连续提取法和碳酸氢盐提取法分析微模型沉积物中铀的化学形态和U(IV)的比例,并利用SEM- EDS和XPS等分析方法分析产物的成分及铀价态.
1 材料和方法
1.1 样品采集与处理
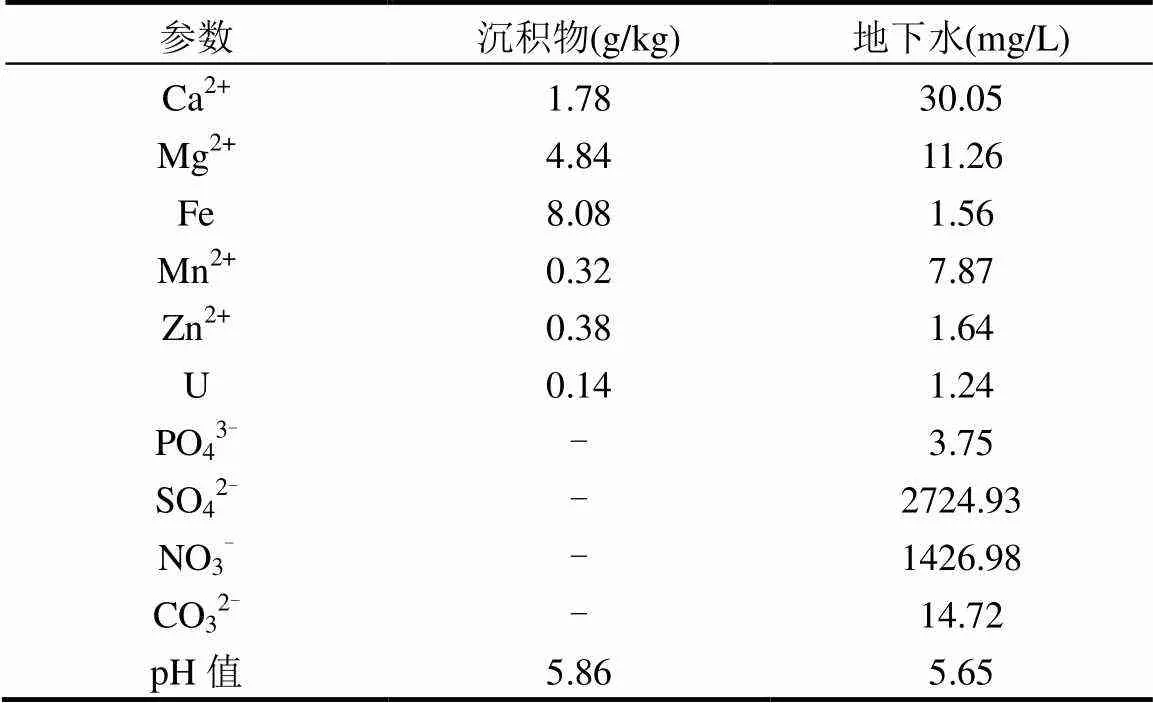
表1 初始沉积物和地下水中的主要理化成分
注:-为未检测.
实验所用地下水和沉积物取自于中国南方某退役铀尾矿库附近.取样点附近地下水中铀浓度为0.5~2.0mg/L,超过了《铀矿冶辐射防护和环境保护规定(GB23727-2009)》[20]规定的排放标准0.050mg/L.地下水样品取自于尾矿库附近一个深约17~18m的监测井,监测井中地下水DO的范围为0.20~0.50mg/L,属于缺氧地下水环境.沉积物取自监测井附近地下5~7m.地下水样与沉积物取样后立即装入经消毒遮光密封的取样瓶和取样袋中,用冰袋包裹立刻送回实验室.用于化学分析的地下水样用0.22μm的滤膜过滤.沉积物和地下水样品中的主要化学成分如表1所示.未经过滤的地下水和剩下的沉积物分别存放在4℃和-20℃的冰箱中备用.
1.2 微模型培养实验
实验分为A、B、C 3组,每组3个平行样.采用1L厌氧水封的广口瓶(并配有12#橡胶塞)作为微模型实验装置.广口瓶消毒灭菌,然后分别加入1L 5mg/L的含铀地下水和100g沉积物,建立沉积物—地下水微模型.A组不做处理作为空白对照,B组加入3g β-甘油磷酸钠,C组加入1.2mL甘油.通过1mol/L HCl或1mol/L NaOH溶液调节微模型中溶液的pH值至6.5,静置过夜.静置过夜后,向微模型中通N2半小时保证微模型的厌氧环境,最后将装置置于厌氧手套箱内25 ℃恒温避光培养.
1.3 铀的分步提取实验
用改良的连续提取方法分析培养结束时沉积物中铀的固相形态.准确称取过100目筛的风干沉积物样品1.0000g置于100mL离心管中,按照如下步骤提取各形态的铀:(1)可交换态:加入15mL 0.4mol/L 的MgCl2,在室温下震荡1h;(2)碳酸盐结合态:往经过(1)处理后的残余物中加入15mL 1mol/L NH4Ac溶液(用HAc调pH值,pH=5),室温搅拌2h;(3)铁锰氧化物结合态:往(2)处理后的残余物中加入15mL 0.1mol/L NH2OH-HCl的25%(/)HAc溶液浸提6h,温度为70℃;(4)有机结合态:往(3)处理后的残余物中加入3mL 0.05mol/L HNO3和5mL 30% H2O2(pH 2~3)在50℃搅拌2h,再加入3mL 30%H2O2(pH 2~3)溶液50℃搅拌2h,冷却后加入5mL 3.2mol/L NH4Ac的20%(/)HNO3溶液,室温下搅拌20min;(5)残渣态:经过(4)处理后的残余物用15mL王水在150℃消解15min.每个提取步骤后,将样品以4000r/min离心20min,取上清液,用0.22μm的滤膜过滤后分析.
1.4 U(IV)提取实验
为了分析沉积物中U(IV)的比例,参照Sharp等[21]提出的方法提取沉积物中的U(IV).具体方法如下:取第25d和第45d的沉积物60℃真空干燥12h,过200目筛,准确称干燥好的沉积物0.1g于50mL离心管中,然后加入10mL除氧后的0.5mol/L NaHCO3(pH 8.2±0.2)溶液,在厌氧条件下125r/min摇动提取10h.离心取上清液,用0.22μm的滤膜过滤后分析.
1.5 检测方法
从添加碳源开始每5d取一次样,分析微模型中硝酸盐、硫酸盐、磷酸盐、pH值和铀浓度的变化.pH值采用pH计测量(HACH,HQ430D,USA);铀浓度采用ICP-MS测量(7700Series,Agilent,USA);硝酸盐、硫酸盐和磷酸盐浓度采用紫外可见分光光度计测量(T6,新世纪,北京);沉积物元素组成用扫描电镜能谱仪分析(FEI,Quanta 250,USA);沉积物中铀的价态用X射线光电子能谱仪分析(Thermo Fisher, ESCALAB 250Xi,USA).
2 结果与讨论
2.1 微模型中硝酸盐、硫酸盐、磷酸盐、pH值、铀浓度的变化
由图1a可以看出:厌氧培养15d左右,B组和C组地下水中硝酸盐浓度由1426.98mg/L下降到7.6mg/L左右,而A组硝酸盐浓度在第5d仅下降到860.68mg/L,之后基本保持不变.地下水中硝酸盐浓度下降的主要原因可能是微生物的反硝化作用[22-23].
图1b中,A组硫酸盐浓度从始至终未发生明显变化,B组硫酸盐浓度在第5d左右开始下降,到第30d时从2.72g/L下降到1.71g/L,从第30~45d浓度基本稳定.C组的硫酸盐浓度变化与B组基本一致.硫酸盐浓度的下降是由于β-甘油磷酸钠/甘油的加入,激活了微生物,特别是激活了硫酸盐还原菌发生异化使硫酸盐还原成硫化氢[24].
从图1c可以看出,A组和C组磷酸盐浓度基本保持不变,而B组微模型中磷酸盐的浓度由初始的3.75mg/L上升到41.16mg/L,并在第10d时达到峰值78.41mg/L,这是由于地下水和β-甘油磷酸钠的化学作用以及环境中微生物的作用,使β-甘油磷酸钠逐渐分解释放磷酸盐,从而使溶液中磷酸盐的浓度升高[6,25].而在第10d后,地下水中的磷酸盐浓度逐渐降低,最终降到25.68mg/L.这可能是由于磷酸盐与溶液中的铀发生矿化作用[19],形成不溶于水的矿化产物,最终使溶液中磷酸盐的浓度下降.
图1d中,在厌氧培养的前5d A、B、C 3组中pH值逐渐上升,这可能是由调节溶液pH值过程中添加的NaOH和HCl与沉积物反应释放了沉积物中的碱性物质,然而B组的pH值高于A、C两组,这可能还与β-甘油磷酸钠水解有关.在第5~45d,B、C两组微模型中pH继续上升并在后期趋于稳定,而A组pH基本保持不变.B、C两组微模型中pH后续的上升可能是由于加入的β-甘油磷酸钠/甘油激活了硫酸盐还原菌群,硫酸盐还原过程会消耗氢离子从而引起微模型中pH值的升高[26].在培养的后期,碳源消耗完全,缺乏碳源的硫酸盐还原菌群停止了硫酸盐还原代谢,pH值也停止上升.
由图1e可知,静置过夜后,地下水中的铀浓度由调节的5.0mg/L下降到3.0mg/L左右,这可能与沉积物对地下水中铀的吸附有关[27-28].厌氧培养5d后,A、B、C 3组微模型中铀浓度均明显下降,之后B、C两组铀浓度继续下降,而A组铀浓度维持在1.70mg/L左右.在厌氧培养后的第5~25d,B组微模型中铀浓度下降速度明显快于C组.在第15d和25d,B组和C组微模型中铀浓度分别下降到最低值0.030mg/L, 0.044mg/L,低于《铀矿冶辐射防护和环境保护规定(GB23727-2009)》的排放标准(<0.050mg/L).但是在第25d后,C组微模型中的铀浓度又开始上升,在第45d时达到0.182mg/L,这可能是因为C组微模型中甘油消耗完毕后微生物耗氧代谢减少导致溶解氧上升,使得还原生成的U(IV)又被氧化成U(VI).而B组微模型中的铀浓度一直保持不变,可能是铀与磷酸盐矿化形成比较稳定的矿化产物[19].

2.2 沉积物中铀的化学形态
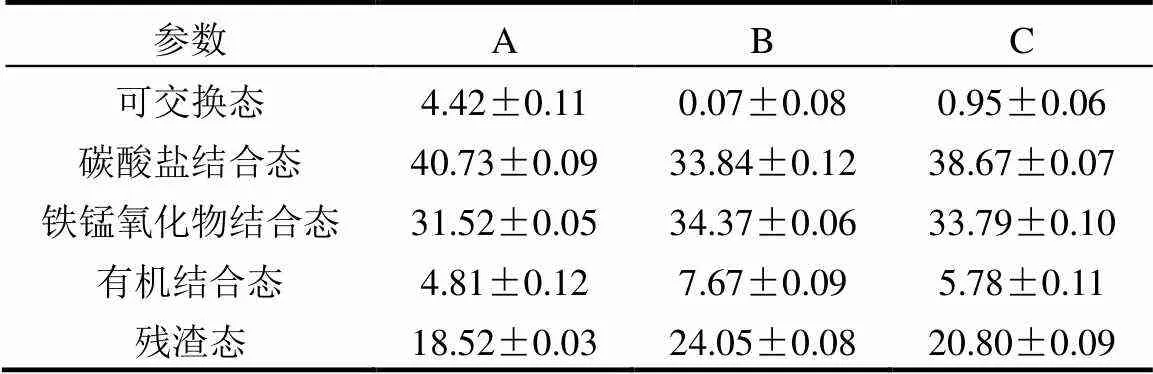
表2 培养至25d时沉积物中各形态铀所占比例(%)
从表2中可以看出:与A组相比B组的可交换态和碳酸盐结合态的比例分别下降了4.35%和6.89%,而铁锰氧化物结合态、有机结合态和残渣态所占的比例分别上升了2.85%、2.83%和5.52%.与C组相比,B组的可交换态和碳酸盐结合态的比例分别下降了0.88%和4.83%,而铁锰氧化物结合态、有机结合态和残渣态所占的比例分别上升了0.58%、1.98%和3.25%.可交换态和碳酸盐结合态的铀在环境中容易受到外界因素的影响而向环境中释放,对环境的潜在威胁很大[29-31].铁锰氧化物结合态、有机结合态和残渣态的铀在自然条件下不易释放能够长期稳定地存在于沉积物中,对环境基本无影响[31-33].β-甘油磷酸钠作为碳源和磷源能够降低沉积物中非稳态的可交换态和碳酸盐结合态铀的比例,并且能提高沉积物中稳定存在的铁锰氧化物结合态、有机结合态和残渣态铀的比例.
2.3 沉积物中U(IV)的比例
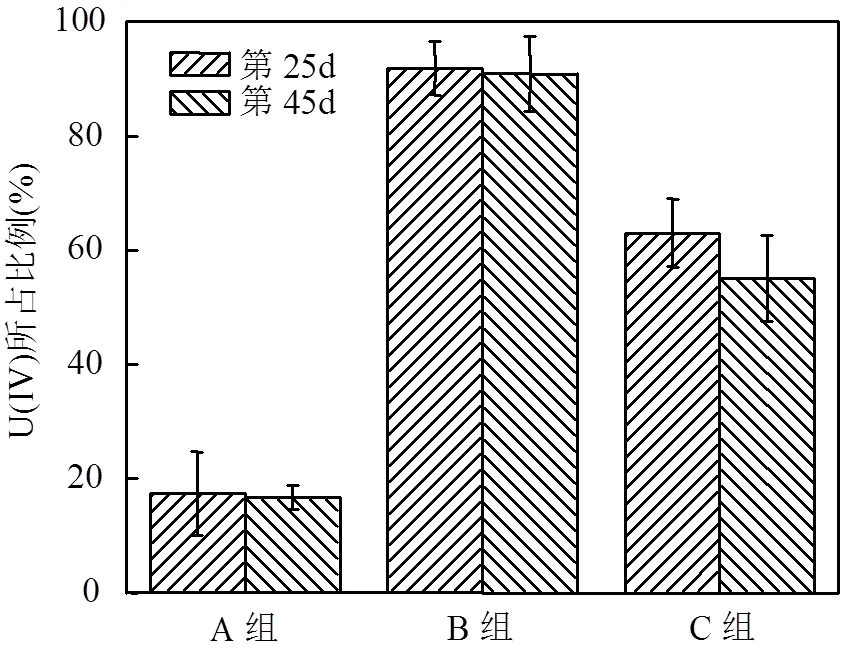
图2 培养至25d和45d时沉积物中U(IV)的比例
由图1e可以看出,在第25d左右,微模型中铀的还原和矿化基本结束,所以选取了第25d和第45d的沉积物,分析沉积物中U(IV)的比例变化.由图2可知,A组第25d和第45d的沉积物中U(IV)的比例基本保持不变,仅占17.36%.这可能是沉积物中本身含有少量的U(IV).C组第25d的沉积物中U(IV)的比例为62.97%,但是在第45d时U(IV)的比例降低到54.99%.这表明甘油作为碳源能促进U(VI)转换成U(IV),但是其还原产物不稳定易被氧化[1].B组第25d的沉积物中U(IV)的比例高达91.79%,且在第45d的沉积物中U(IV)的比例没有明显下降.这说明β-甘油磷酸钠作为碳源和磷源促进了U(VI)转换成U(IV),形成的产物相对C组比较稳定不易被氧化[25].

图3 β-甘油磷酸钠组培养至25d时沉积物的SEM-EDS
2.4 扫描电镜/能谱分析
从图3中可以看出,沉积物中含有磷、铀、钙、氧、硅、铝、铁等元素.磷、铀和钙3种元素的赋存位置基本保持一致,且出现了较为明显的磷、铀和钙的信号峰,说明β-甘油磷酸钠作为碳源和磷源修复含铀地下水过程中可能生成了一种含Ca、U、P的难溶产物[6].
2.5 X射线光电发射光谱分析
由图4a XPS全元素扫描谱中可以看出,沉积物中含有磷、铀、钙、氧、硅、铝和铁等元素.从图4b可以看到,在结合能390.54,379.73eV处分别出现2组峰U4f7/2和U4f5/2,且两峰之间的电位差约为10.81eV.分峰处理后,U4f7/2轨道处的峰由U(IV)在379.73eV处的峰和U(VI)在381.82eV处的峰叠加而成,其含量比约为9:1;U4f5/2轨道处的峰由U(IV)在390.54eV处的峰和U(VI)在391.97eV处的峰叠加而成,其含量比约为9:1[34].由此可知,β-甘油磷酸钠处理后第25d沉积物中含有约90%的U(IV),而这与之前U(IV)提取实验得出的结果基本一致.
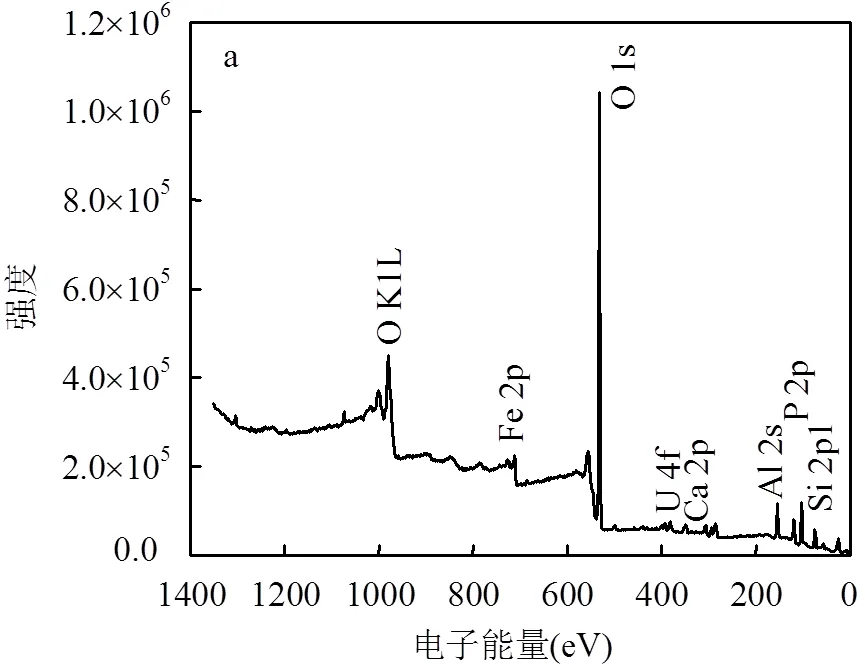
a. XPS全元素扫描图谱; b. U4f 谱图
3 结论
3.1 β-甘油磷酸钠作为碳源和磷源能够激活土著微生物发生酶促反应,将U(VI)转换成U(IV)从而将铀原位固定,使地下水中铀的浓度在第25d时降到0.030mg/L,达到了《铀矿冶辐射防护和环境保护规定(GB23727-2009)》的排放标准(<0.050mg/L).
3.2 厌氧培养结束后,连续提取法对沉积物中铀的固相形态分析结果表明:与甘油组相比,可交换态和碳酸盐结合态的铀的比例分别降低了0.88%和4.83%,稳定存在的铁锰氧化物结合态、有机结合态和残渣态的铀的比例分别提高了0.58%、1.89%和3.25%,且生成的产物相对较稳定不易被氧化.
3.3 沉积物中U(IV)提取实验和光谱分析结果表明沉积物中大约有91.72%的U(VI)被转化成U(IV).
[1] Newsome L, Morris K, Lloyd J R. The biogeochemistry and bioremediation of uranium and other priority radionuclides [J]. Chemical Geology, 2014,363(1):164-184.
[2] 稂 涛,胡 南,张 辉,等.博落回对不同化学形态铀的富集特征[J]. 环境科学研究, 2017,(8):1238-1245.
[3] Mccarthy J F, Yurek G J, Elliott J F, et al. Update on the chemical thermodynamics of uranium, neptunium, plutonium, americium and technetium [J]. The American Industrial Hygiene Association Journal, 1982,(43):880-886.
[4] 吴唯民,Carley Jack, Watson David,等.地下水铀污染的原位微生物还原与固定:在美国能源部田纳西橡树岭放射物污染现场的试验[J]. 环境科学学报, 2011,31(3):449-459.
[5] 孙 静,丁德馨,胡 南,等.诱变绳状青霉吸附铀的行为研究[J]. 环境科学学报, 2012,32(10):2384-2393.
[6] 胡 南,李 科,戴仲然,等.黑曲霉溶解磷矿粉矿化水体中铀的研究 [J]. 铀矿冶, 2017,(4):289-295.
[7] 聂小琴,丁德馨,李广悦,等.某铀尾矿库土壤核素污染与优势植物累积特征[J]. 环境科学研究, 2010,(6):719-725.
[8] Luo W, Gu B. Dissolution of uranium-bearing minerals and mobilization of uranium by organic ligands in a biologically reduced sediment [J]. Environmental Science & Technology, 2011,45(7):2994- 2999.
[9] 魏广芝,徐乐昌.低浓度含铀废水的处理技术及其研究进展[J]. 铀矿冶, 2007,26(2):90-95.
[10] 谭文发,吕俊文,唐东山.生物技术处理含铀废水的研究进展 [J]. 生物技术通报, 2015,31(3):82-87.
[11] Begg J D C, Burke I T, Lloyd J R, et al. Bioreduction behavior of U(VI) sorbed to sediments [J]. Geomicrobiology Journal, 2011,28(2): 160-171.
[12] Lovely D R, Phillips E J P, Gorby Y A, et al. Microbial uranium reduction [J]. Nature, 1991,350:413-416.
[13] Williams K H, Bargar J R, Lloyd J R, et al. Bioremediation of uranium-contaminated groundwater: a systems approach to subsurface biogeochemistry [J]. Current Opinion in Biotechnology, 2013,24(3): 489-497.
[14] Wan J, Tokunaga T K, Brodie E, et al. Reoxidation of bioreduced uranium under reducing conditions [J]. Environmental Science & Technology, 2005,39(16):6162-6169.
[15] Law G T W, Geissler A, Burke I T, et al. Uranium redox cycling in sediment and biomineral systems [J]. Geomicrobiology Journal, 2011,28(5/6):497-506.
[16] Mehta V S, Maillot F, Wang Z, et al. Transport of U(VI) through sediments amended with phosphate to induce in situ uranium immobilization [J]. Water Research, 2015,69:307-317.
[17] Pinto A J, Gonçalves M A, Prazeres C, et al. Mineral replacement reactions in naturally occurring hydrated uranyl phosphates from the Tarabau deposit: Examples in the Cu–Ba uranyl phosphate system [J]. Chemical Geology, 2012,s312–313(3):18-26.
[18] Wellman D M, Icenhower J P, Owen A T. Comparative analysis of soluble phosphate amendments for the remediation of heavy metal contaminants: effect on sediment hydraulic conductivity [J]. Environmental Chemistry, 2006,3(3):219-224.
[19] Newsome L, Morris K, Trivedi D, et al. Biostimulation by glycerol phosphate to precipitate recalcitrant uranium(IV) phosphate [J]. Environmental Science & Technology, 2015,49(18):11070-8.
[20] GB 23727-2009 铀矿冶辐射防护和环境保护规定[s].
[21] Sharp J O, Lezama-Pacheco J S, Schofield E J, et al. Uranium speciation and stability after reductive immobilization in aquifer sediments [J]. Geochimica Et Cosmochimica Acta, 2011,75(21):6497- 6510.
[22] Suzuki Y, Kelly S D, Kemner K M, et al. Microbial populations stimulated for hexavalent uranium reduction in uranium mine sediment [J]. Applied & Environmental Microbiology, 2003,69(3): 1337-1346.
[23] Moon H S, Mcguinness L, Kukkadapu R K, et al. Microbial reduction of uranium under iron- and sulfate-reducing conditions: Effect of amended goethite on microbial community composition and dynamics [J]. Water Research, 2010,44(14):4015-4028.
[24] Luo J, Weber F A, Cirpka O A, et al. Modeling in-situ uranium(VI) bioreduction by sulfate-reducing bacteria [J]. Journal of Contaminant Hydrology, 2007,92(1/2):129-148.
[25] Salome K R, Green S J, Beazley M J, et al. The role of anaerobic respiration in the immobilization of uranium through biomineralization of phosphate minerals [J]. Geochimica Et Cosmochimica Acta, 2013,106(106):344-363.
[26] Zagury G J, Kulnieks V I, Neculita C M. Characterization and reactivity assessment of organic substrates for sulphate-reducing bacteria in acid mine drainage treatment [J]. Chemosphere, 2006,64(6): 944-954.
[27] Camacho L M, Deng S, Parra R R. Uranium removal from groundwater by natural clinoptilolite zeolite: Effects of pH and initial feed concentration [J]. Journal of Hazardous Materials, 2010,175(1-3): 393-398.
[28] Fassett J E, Heaman L M, Simonetti A. Direct U-Pb dating of Cretaceous and Paleocene dinosaur bones, San Juan Basin, New Mexico Reply [J]. Geology, 2012,40(4):E263-E264.
[29] Rout S, Kumar A, Ravi P M, et al. Understanding the solid phase chemical fractionation of uranium in soil and effect of ageing [J]. Journal of Hazardous Materials, 2016,317:457-465.
[30] Echevarria G, Sheppard M I, Morel J. Effect of pH on the sorption of uranium in soils [J]. Journal of Environmental Radioactivity, 2001, 53(2):257-264.
[31] Vandenhove H, Vanhoudt N, Duquène L, et al. Comparison of two sequential extraction procedures for uranium fractionation in contaminated soils [J]. Journal of Environmental Radioactivity, 2014, 137:1-9.
[32] Filgueiras A V, Lavilla I, Bendicho C. Chemical sequential extraction for metal partitioning in environmental solid samples [J]. Journal of Environmental Monitoring, 2002,4(6):823-857.
[33] Schultz M K, Burnett W C, Inn K G W. Evaluation of a sequential extraction method for determining actinide fractionation in soils and sediments [J]. Journal of Environmental Radioactivity, 1998,40(2): 155-174.
[34] Li X, Liao J, Liang D, et al. Microbial reduction of uranium (Ⅵ) by Bacillus sp.dwc-2:A macroscopic and spectroscopic study [J]. Journal of Environmental Sciences, 2017,53(3):9-15.
Remediation of uranium contaminated groundwater by β-glycerophosphate.
HUANG Chao1,2, ZHANG Hui1,2, HU Nan1,2, DING De-xin1,2*, LI Dian-xin1,2, SUN Jing1,2
(1.Key Discipline Laboratory for National Defense for Biotechnology in Uranium Mining and Hydrometallurgy, University of South China, Hengyang 421001, China;2.Hunan Province Key Laboratory of Green Development Technology for Extremely Low Grade Uranium Resources, University of South China, Hengyang 421001, China)., 2018,38(9):3391~3397
The microcosm experiment were conducted to investigate the remediation effect of uranium contaminated groundwater by β-glycerophosphate. The variations of pH and nitrate, sulfate, phosphate, and uranium concentrations were monitored during the incubation, and the uranium species and the proportion of U(IV) in sediments were analysed. It was found that, after 25d incubation, the concentrations of uranium in liquids in the β-glycerophosphate and glycerol amended groups decreased from 2.96 and 2.99mg/L to 0.030 and 0.044mg/L, respectively, which were lower than the limits of contaminants stipulated in the regulations on radiation and environment protection in uranium mining and milling in China. β-glycerophosphate worked as the carbon and phosphate sources and reduced the proportion of the uranium species in exchangeable and carbonate fractions in sediments and increased the proportion of the uranium species in Fe(Mn) oxides, organic matter and residual fractions in sediments. The proportion of U(IV) in the sediment of β-glycerophosphate amended group amounted to 91.79%, which was 28.82% higher than that of the glycerol amended group. After 45d anaerobic incubation, the proportion of U(IV) in the sediment of β-glycerophosphate amended group almost remained unchanged, while that in glycerol amended group decreased by 7.98%. Therefore, β-glycerophosphate could work as carbon and phosphate sources and could immobilize uranium in-situ through bioreduction and biomineralization.
β-glycerophosphate;uranium contaminated groundwater;bioreduction;biomineralization;in-situ immobilization
X52
A
1000-6923(2018)09-3391-07
黄 超(1993-),男,湖南益阳人,南华大学硕士研究生,主要从事铀污染地下水生物修复研究.
2018-02-08
国家自然科学基金重点项目(U1401231);国家自然科学基金面上项目(11775106);湖南省教育厅一般项目(15C1179)
* 责任作者, 教授, dingdxzzz@163.com
